Chapitre
III :
Etude
de la structuration de peptides sur support solide par RMN HRMAS
1 La synthèse peptidique
1.1 Synthèse peptidique en phase homogène
Avant l’apparition de la synthèse en phase solide, la synthèse peptidique était réalisée en phase homogène. Bien que reposant sur une chimie simple, avec la formation d’une succession de liens amides, la synthèse peptidique en phase homogène n’est pas si facile à réaliser. Les premières synthèses de peptides en phase homogène consistaient en une suite de condensation de fragments. Un octapeptide était synthétisé à partir de deux tétrapeptides eux-même obtenus à partir de deux dipeptides. Une autre alternative à la méthode utilisant des couplages entre différents fragments, consistait en une succession de couplages sur la chaîne peptidique en croissance. La méthode de synthèse pas à pas a été utilisée pour la première fois avec succès par Du Vigneau et al lors de la synthèse de l’ocytocine, peptide de 9 acides-aminés.[1] Lors du couplage de deux acides aminés, on n’obtient pas seulement la molécule désirée mais un mélange de molécules. On obtient le dipeptide désiré mais aussi le dipeptide inverse ainsi que des polymères. Chaque étape de couplage nécessite donc une étape de purification. La synthèse de peptides de tailles importantes par cette méthode, devient vite fastidieuse car les différentes étapes de purification entre chaque couplage prennent beaucoup de temps. Afin de faciliter les étapes de purification, une méthode basée sur des esters activés a été développée.[2] Cette méthode conduisait à des mélanges de molécules moins complexes, puisque le recours à des esters activés permet de favoriser la réaction désirée. Un autre problème de la synthèse peptidique en phase homogène est que ce mode de synthèse ne permet pas l’utilisation d’un grand excès de réactifs pour conduire la réaction jusqu'à son terme car tout excès de réactifs doit être éliminé et cela nécessite une étape de purification.
1.2 Synthèse peptidique en phase solide.
Depuis son introduction en 1963 par Merrifield, la synthèse en phase solide a considérablement reculé les limites de la synthèse peptidique connues en phase homogène. Merrifield a eu l’idée de combiner la méthodologie utilisant les esters activés avec l’ancrage du premier acide aminé sur une matrice polymère insoluble. Ce nouveau mode de synthèse permettait donc d’éviter les réactions indésirables puisque avec l’immobilisation d’un des partenaires sur la phase solide, une seule face d’un acide aminé est disponible pour le couplage. Un autre avantage de l’immobilisation d’un acide aminé sur un polymère insoluble est que cela permet l’utilisation d’un excès du réactif en solution. L’excès de réactif n’est plus considéré comme une impureté pour le produit final et permet d’obtenir des rendements de couplage proche de 100%. L’avantage sûrement le plus conséquent de la synthèse en phase solide, est que toutes les étapes de purification entre chaque couplage sont supprimées puisque le produit désiré reste accroché sur le polymère insoluble, jusqu'à obtention du peptide désiré. Le peptide est ensuite séparé du polymère par coupure chimique. L’avancée réalisée par la chimie peptidique en phase solide a contribué au développement de nombreux supports solides, ainsi qu’au développement de nouvelles chimies pour l’activation et la protection des acides aminés. Actuellement deux grandes stratégies sont utilisées pour la synthèse peptidique en phase solide. Ces deux stratégies reposent sur l’utilisation de deux types de protections temporaires dites orthogonales Fmoc/Boc (schéma 3), pour la protection de la fonction aminea.
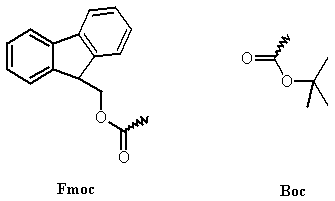
Schéma 3 : Formules développées planes des groupements Fmoc (9-fluorénylmethyloxycarbonyle) et Boc (tertio-butyloxycarbonyl).
La différence principale entre les deux types de groupes protecteurs est liée à leur mode de déprotection. La déprotection du groupement Fmoc s’effectue en milieu basique alors que la déprotection du groupement Boc s’effectue en milieu acide. Actuellement la stratégie Fmoc/tBu est la plus utilisée car la coupure finale du peptide du support solide utilise du TFA concentré alors que la stratégie Boc/Bnl nécessite l’utilisation d’acide fluorhydrique concentré. Bien que ces deux types de protection se déprotègent selon deux protocoles différents, le protocole utilisé pour le couplage des différents acides aminés est le même. La synthèse peptidique en phase solide consiste donc en une succession de couplages et de déprotections jusqu’à obtention du peptide désiré qui sera ensuite séparé du polymère par coupure chimique (figure 19). Lors de la synthèse de peptides en phase solide, les seules techniques qui permettent d’évaluer l’efficacité d’un couplage ou d’une déprotection reposent sur des tests colorimétriques qui ne sont pas quantitatifs.[3], [4] Pour pouvoir analyser la chaîne peptidique de façon quantitative il faut séparer la molécule du support solide pour ensuite l’analyser avec les techniques habituelles RMN, HPLC et la spectroscopie de masse. Ce procédé, qui consiste à couper la molécule du support avant de pouvoir l’analyser, n’est pas satisfaisant dans l’optique de comprendre les phénomènes qui sont à l’origine des problèmes rencontrés lors de la synthèse de séquences dites difficiles.[5], [6] En dehors des séquences difficiles, la synthèse peptidique en phase solide pose un problème général pour la synthèse de peptides longs. Les rendements de couplages approchent en général 100%. Pour la synthèse d’un peptide de 50 acides aminés par exemple un rendement par couplage de 99% conduit à une perte de peptide importante et la chute du rendement global devient dramatique des lors que le rendement par couplage est de 95 %(tableau 2). Ce phénomène s’accentue au fur et a mesure que la longueur de la séquence augmente
|
|
Longueur de séquence |
10 |
50 |
100 |
|
Rendement par couplage |
|
|
|
|
|
90% |
|
34 |
0,5 |
0,002 |
|
95% |
|
59 |
8 |
0.6 |
|
99% |
|
90 |
60 |
36 |
|
99,5% |
|
95 |
77 |
60 |
|
99,9% |
|
99 |
95% |
90 |
Tableau 2 : évolution du rendement global de synthèse en fonction de la longueur du peptide et du rendement par couplage.
En marge des peptides longs, certaines séquences peptidiques posent des problèmes dès les premiers acides aminés. Ces séquences dites difficiles sont des séquences caractérisées par des couplages et/ou des déprotections incomplètes. Ces problèmes ont souvent été attribués à une structuration de la chaîne peptidique ou encore à une mauvaise solvatation. Peu de techniques permettent de déterminer si ces phénomènes sont réellement à l’origine des problèmes rencontrés pour les séquences difficiles.

Figure 19 : Principe de la synthèse peptidique en phase solide.
L’IR-FT ou encore la RPE peuvent apporter un élément de réponse mais ces techniques nécessitent une modification de l’échantillon. L’IR-FT utilise des acides-aminés deutérés, et la RPE nécessite l’emploi d’un groupement paramagnétique. L’emploi de ces techniques est donc très contraignant et n’est pas toujours facile à mettre en œuvre. En 1997, notre groupe a montré que la RMN HRMAS permettait de suivre la croissance de la chaîne peptidique lors de la synthèse d’une séquence dite difficile.[7] Lors de cette étude, il a été montré que les problèmes rencontrés lors de la synthèse de l’aggrécan étaient liés à des problèmes de solvatation des chaînes. Cette même étude a montré une corrélation directe entre la mobilité des chaînes de peptides et la qualité des spectres obtenus. La RMN HRMAS montre donc de grandes possibilités en ce qui concerne l’étude de la chaîne peptidique lorsque celle-ci est encore ancrée sur le support. Néanmoins, la question de la structuration des peptides sur le support solide n’est pas encore résolue. Notre groupe a déjà été impliqué dans la détermination de la structure secondaire de peptides lorsque ceux-ci étaient encore ancrés sur le support solide. L’utilisation des deux chimies orthogonales a permis de mettre en évidence que des peptides ayant une forte tendance à l’agrégation pouvaient adopter une structure secondaire sur la résine[8]. Dans ce cas, la résine était utilisée comme « chaperonne chimique ». En effet, l’utilisation simultanée de la chimie Boc et de la chimie Fmoc a permis de diluer le peptide sur la résine à chaque fois que la qualité du spectre RMN HRMAS se détériorait. Lors de cette étude, il a été montré que l’homo-polymère d’alanine, à partir d’une certaine longueur (12 résidus), pouvait adopter une structure en hélice alpha sur le support solide, à conditions que les chaînes ne soient pas agrégées aux étapes antérieures. Toutefois, cette étude avait été menée dans un solvant organique incompatible avec l’étude de peptides d’intérêt biologique qui nécessitent un milieu aqueux. Cette utilisation de la résine comme chaperonne chimique pourrait également servir dans l’étude de certains peptides qui agrégent in vivo, peptides qui sont à la source de plusieurs pathologies comme la maladie d’Alzheimer. Les phénomènes à l’origine de l’agrégation des ces molécules sont encore mal connus et différentes hypothèses sont avancées. Une des premières hypothèses est le passage d’une forme random coil soluble à une forme structurée en feuillet béta intermoléculaire moins soluble. La deuxième hypothèse avancée est une augmentation de la concentration locale lors du vieillissement des cellules.[9] La compréhension des phénomènes qui sont à l’origine de l’agrégation des peptides et protéines est donc cruciale. Nous avons évalué la RMN HRMAS combinée à la synthèse en phase solide pour tester d’abord la compatibilité des différents supports hydro-gonflables avec une structuration en structure secondaire. En effet, dans le cas où la matrice polymère ne serait pas un obstacle à la structuration de peptides ou de protéines, l’immobilisation d’un objet d’intérêt biologique sur un support solide pourrait être mise à profit lors de l’étude de sa structuration avant qu’il n’agrège.
2 Etude de la structuration d’un peptide sur différents supports solides
2.1 Caractéristiques RMN des deux principales structures secondaires
2.1.1 Hélice alpha
L’hélice alpha est l’une des deux principales structures secondaires rencontrées dans les peptides et protéines. Chacune de ces structures secondaires est caractérisée par une « signature RMN ». Dans un premier temps, la détermination de la structure secondaire des peptides et protéines passe par l’attribution séquentielle de la chaîne peptidique. De l’attribution séquentielle, on peut déjà tirer une première information concernant la structure secondaire des peptides et protéines. En utilisant la variation des déplacements chimiques par rapport à leur valeur dans un peptide non structuré (random coil), on peut déjà obtenir une idée de la structuration du peptide.[10],[11],[12] L’étude de la déviation des déplacements chimiques par rapport à la valeur random coil a déjà était adaptée à l’étude de peptides sur le support solide[13], gonflés dans un solvant organique, permettant ainsi de ré-établir l’échelle du random coil. Dans le cas d’une hélice alpha, les déplacements chimiques des protons alpha subissent une déviation vers les hauts champs et connaissent une faible dispersion. La détermination des constantes des couplages, donne des informations sur les angles de torsion, ce qui permet également d’obtenir des informations sur la structure de la chaîne peptidique. L’hélice alpha est caractérisée par des valeurs 3JHNa de l'ordre de 3,9Hz, correspondant à un angle f de –57°. L’autre caractéristique de la structuration d’un peptide provient de l’analyse des contacts NOE.[14] Les hélices alpha sont caractérisées par des contacts NH-NH forts ainsi que des contacts ab (i, i+3). Il est à noter que l’hélice a est stabilisée par des ponts hydrogènes entre les N-H et les C=O des résidus i et i+3.
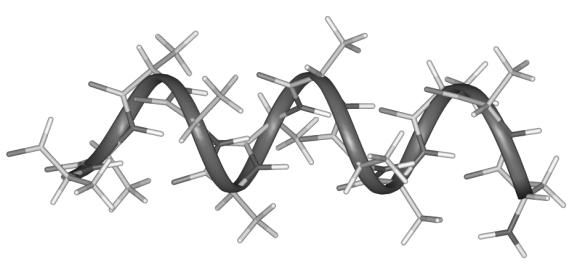
Figure 20 : Modélisation moléculaire a l’aide du logiciel INSIGHT II (V2000.1 Accelrys Inc.) d’un peptide poly-ala12 formant une hélice alpha.
2.1.2 Feuillet béta
Le feuillet béta est l’autre type de structure secondaire rencontrée dans les peptides et protéines et est constitué de deux ou plusieurs fragments étendus, reliés entre eux par une région formant un coude. Le feuillet béta possède souvent une plus forte tendance à l’agrégation intermoléculaire que l’hélice alpha car ses fonctions N-H et C=O ne sont pas impliquées dans des ponts hydrogènes avec des résidus voisins dans la séquence primaire. Comme l’hélice alpha, le feuillet béta est caractérisé par une « signature RMN ». Concernant la déviation des déplacements chimique par rapport à la valeur random coil, les protons alpha des acides aminés constituant le feuillet sont déviés à bas champs (grandes valeurs de ppm). Concernant les constantes de couplages, les feuillets béta sont caractérisés par une constante 3JHNa d'environ 9,7 Hz correspondant à un angle de torsion f de –119°. Dans le cas des contacts NOE, les feuillets béta parallèles sont caractérisés par des contacts aN (i,i+1) forts. D’une façon plus importante, des contacts NOEs entre les différents brins permettent de distinguer le feuillet b d’une simple structure étendue. Des contacts Hai-Haj, où i et j représentent deux résidus sur les différents brins, ainsi que des contacts entre chaînes latérales de résidus éloignés dans la séquence primaire, sont caractéristiques. Finalement la RMN permet en général de définir la région du tournant, avec un contact HNi-HNi+1 entre les deux résidus centraux où la chaîne se retourne.
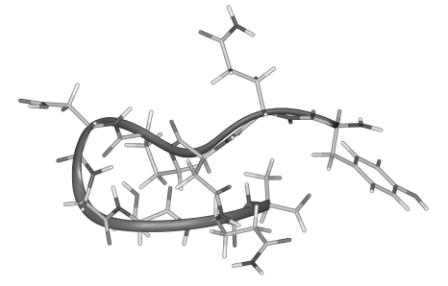
Figure 21 : Modélisation moléculaire a l’aide du logiciel INSIGHT II (V2000.1 Accelrys Inc.) du peptide YQNPDGSQA formant un béta hairpin.
2.2 Etude d’un peptide formant un feuillet béta sur différentes résines.
De nombreux peptides ont été proposés pour aborder la formation de feuillets béta intramoléculaires en solution. Néanmoins, le succès utilisant des acides aminés naturels est resté limité, contrairement au modèle de l’hélice alpha. Le problème vient de la nature non locale des interactions et de la tendance à favoriser l’association intermoléculaire plutôt que la formation d’un feuillet intramoléculaire. L’ajout de co-solvants organiques peut diminuer cette tendance à l’agrégation, mais n’est pas satisfaisant lors de l’étude de peptides à visée biologique. Il est clair que la région centrale du feuillet impliquant le tournant béta, joue un rôle clé. Afin d’imposer le retour correct de la chaîne peptidique, des mimes organiques rigides tels que le dibenzofurane (DBF) ont été proposés.[15],[16] Néanmoins certains peptides naturels semblent adopter au moins partiellement une structure en feuillet béta monomérique en solution. Nous avons choisi un tel peptide, dérivé de la protéine tendamistat, ayant la séquence YQNPDGSQA.[17] Ce peptide montre en solution dans l’eau à 4°C les caractéristiques de deux brins étendus, d’une région charnière centrée autour des résidus Asp Gly, et surtout quelques contacts NOE inter-brins. La question était de voir si ce même peptide greffé sur différents supports hydro-gonflables allait aussi adopter la même conformation. Pour valider la compatibilité de la structuration avec le support solide, nous nous sommes tout d’abord consacrés à l’étude de ce peptide modèle de feuillet béta intramoléculaire.
2.2.1 Formation d’un feuillet béta sur Péga-HMBA ?
Dans un premier temps nous avons synthétisé le peptide sur la résine péga-HMBA chargée à 0,38 mmol/g. Cette résine est une résine amino péga, fonctionnalisée par le linker de Sheppard. Les résines de type péga sont des résines constituées de diméthyl acrylamide et de polyéthylène glycol.
Schéma 4 : Formule développée plane de la résine Péga-HMBA fonctionnalisée par le linker de Sheppard.
Le PEG a déjà montré sa compatibilité avec les protéines. La ligation de PEG avec des protéines permet d’une part de réduire la tendance à l’agrégation mais permet aussi d’augmenter leur solubilité.[18] Le PEG a un effet non dénaturant, puisque les protéines liées au PEG conservent leur structure native et il a aussi été montré que les enzymes conservaient leur activité catalytique en présence de PEG.[19] Nous avons porté notre choix sur cette résine de part sa compatibilité avec les milieux aqueux, mais aussi pour son ancrage résistant au TFA lors de la déprotection des chaînes latérales des différents acides aminés. Le peptide a été synthétisé selon la stratégie Fmoc/Tbu sur une échelle de 0,1 mmol. L’acquisition des spectres RMN a été réalisée sur un spectromètre Bruker DMX à 600MHz. Les spectres ont été enregistrés dans les conditions décrites par Blanco et al12, à savoir 4°C dans un tampon phosphate à pH 5.
Dans un premier temps, l’attribution des différentes résonances a pu se faire par analyse du spectre TOCSY. Sur le TOCSY, toutes les résonances ont pu être identifiées et les déplacements chimiques des protons alpha subissent une déviation vers les bas champs par rapport à la valeur random coil. Cette déviation des déplacements chimiques des Ha vers les bas champs est une bonne indication de la structuration possible du peptide en feuillet béta. De plus, les déplacements chimiques observés pour le peptide sur la résine sont proches de ceux observés pour le peptide en solution (figure 22).
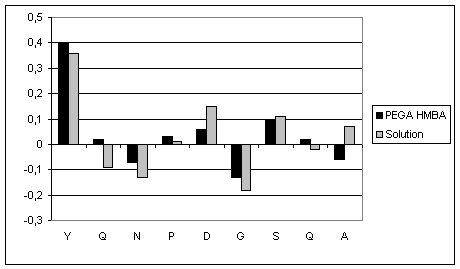
Figure 22 : Comparaison des déplacements chimiques des protons alpha pour le peptide en solution, et sur Péga HMBA, avec la valeur random coil.
En plus de l’analyse de la déviation des déplacements chimiques des Ha par rapport à la valeur random coil, nous avons aussi utilisé les valeurs des déplacements chimiques des carbones alpha du peptide. L’attribution de l’HSQC 13C nous a permis de déterminer les valeurs des déplacements chimiques des Ca (Figure 24) et nous avons ensuite comparé nos valeurs à celles des mêmes résidus dans un peptide non structuré. Le graphique résultant de la comparaison de nos valeurs avec les valeurs du CSI (Chemical shift Index, valeurs des déplacements chimiques dans un peptide non structuré) est également en bon accord avec la structuration du peptide en feuillet béta sur la résine (figure 23).
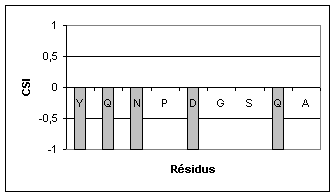
Figure 23 : Index des déplacements chimiques des carbones alpha des 9 résidus du peptide b-hairpin greffé sur Péga-HMBA.

Figure 24 : Spectre HSQC refocalisé de la peptidyl résine enregistré à 4°C dans un tampon phosphate à pH 5 (réel 3,5), montrant l’attribution des Ca du peptide.
Comme nous l’avons vu dans le paragraphe précédent concernant la « signature RMN » des différents structures secondaires, les feuillets béta sont caractérisés par des contacts aN (i, i+1) forts. Nous avons donc procédé à une analyse détaillée du spectre NOE afin de déterminer avec certitude que le peptide adopte au moins une fraction du temps une conformation en feuillet béta intramoléculaire. L’analyse qualitative du spectre NOE montre des contacts aN (i, i+1) forts, mais aussi un contact NH-NH D5->G6, indiquant l’inversion de la chaîne peptidique, ainsi qu’un contact longue distance entre les protons béta de l’alanine avec les protons delta de la tyrosine (Figure 25 et 26).
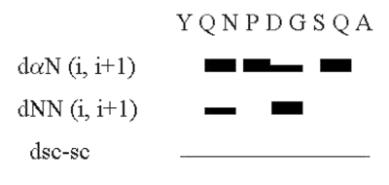
Figure 25 : Résumé des contacts NOE caractéristiques de la formation d’un feuillet béta, sur la résine Péga-HMBA 0,38 mmol/g.
La somme de ces contacts nous permet d’établir avec certitude que le peptide adopte, au moins une fraction du temps, une conformation en feuillet béta intramoléculaire sur la résine péga HMBA.
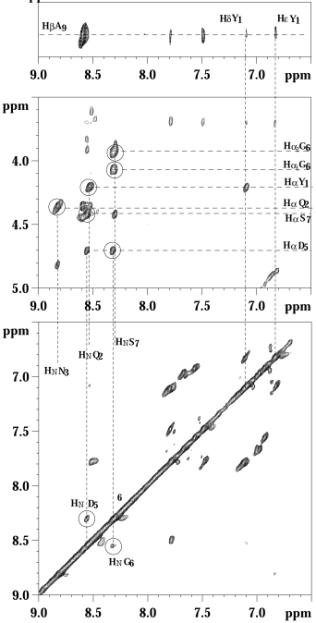
Figure 26 : Superposition de trois zones d’intérêts du spectre HRMAS NOESY (4°C pH 5) du peptide YQNPDGSQA sur péga-HMBA
La structuration d’un peptide en feuillet béta intramoléculaire semble donc être compatible avec la présence d’un support solide. Afin de déterminer si la structure du polymère avait une influence sur la structuration du peptide, nous avons étudié la même séquence sur d’autres supports solides.
2.2.2 Formation d’un feuillet béta sur POEPOP ?
Pour vérifier si la structure du polymère utilisé pouvait influencer la structuration de notre peptide, nous avons utilisé une résine uniquement basée sur du polyéthylène glycol. La résine POEPOP est un polymère de poly-oxy-éthylène et de poly-oxypropylène.

Schéma 5 : Formule développée plane de la résine POEPOP
Cette résine a déjà montré ses qualités en terme de largeur de lignes pour les spectres RMN HRMAS lors de précédentes études.[20],[21] Nous avons donc synthétisé le peptide sur la résine POEPOP, puis nous avons procédé à son analyse RMN HRMAS dans les mêmes conditions que pour la résine Péga-HMBA. Dans un premier temps, la qualité des spectres TOCSY et NOESY que nous avons obtenus était vraiment désastreuse en terme de largeur de ligne. De plus, le spectre NOESY ne comportait que très peu de contacts. Ce résultat était vraiment surprenant vue la qualité des spectres obtenus sur cette résine dans d’autres études.
Après plusieurs tentatives, avec la même résine, nous avons compris que la seule différence, hormis la résine elle-même, était la façon dont l’échantillon avait été préparé pour l’analyse RMN. Dans le cas de la résine POEPOP, le pH du surnageant de la peptidyl résine a été ajusté à 5 alors que dans le cas de la résine Péga-HMBA, la résine a été simplement gonflée dans un tampon phosphate à pH 5. Nous avons donc attribué cet échec à une différence de pH entre les deux échantillons. Afin de vérifier si l’hypothèse d’une différence de pH entre les deux échantillons était la cause des problèmes constatés pour le peptide sur POEPOP, nous avons re-préparé un échantillon de Péga dans les mêmes conditions que précédemment et le pH de la solution surnageante a été contrôlé. Après mesure du pH, nous avons obtenu une valeur de 3,5. Cette chute du pH est probablement due a la présence des contre-ions TFA obtenus lors de la déprotection du peptide. Un nouvel échantillon de peptide sur POEPOP a été préparé en tenant compte de la nouvelle valeur de pH. Les résultats RMN montrent comment ce paramètre peut être déterminant. En effet, le simple passage de pH 5 à pH 3,5 pour la résine POEPOP améliore considérablement la résolution des spectres. De plus, l’attribution du spectre TOCSY présente les mêmes déplacements chimiques que dans le cas de la résine Péga (figure 27) et le spectre NOESY présente les mêmes contacts caractéristiques de la structuration du peptide en feuillet béta intramoléculaire (figure 28). Une première interprétation possible de l’influence du pH est son rôle dans le gonflement des résines. Nous avons utilisé le protocole de la seringue pour effectuer ces différentes mesures.[22] Pour les deux peptidyl résines Péga et POEPOP, le gonflement était supérieur pour les pH bas. Par contre, le gonflement était uniforme quel que soit le pH en absence de peptide. Pour la résine POEPOP supportant le peptide, le volume de gonflement ramené à 1 g de résine passe de 8,7 ml à 11,6 ml en diminuant le pH de 5 à 3,5. Cette même évolution est constatée pour la peptidyl résine Péga : le volume de 1 g de résine passe de 8,7 ml à 9,24 ml pour la même évolution du pH. Dans le cas de l’étude de l’aggrécan, une corrélation directe avait été établie entre une diminution du volume de gonflement de la résine et la qualité des spectres, et l’addition d’un co-solvant polaire tel que le DMSO avait permis de restaurer la qualité des spectres. Ces résultats avaient suggéré que l’agrégation du peptide conduisait à une réticulation supplémentaire non covalente de la résine. Dans le cas de notre peptide, le pI calculé est de l’ordre de 5,1. Cette valeur combinée à la grande densité de chaînes peptidiques sur la résine est probablement à l’origine des problèmes rencontrés. À pH 5, une agrégation même temporaire des chaînes empêche un bon gonflement et mène vers une qualité médiocre des spectres. Descendre le pH à une valeur de 3,5, réduit cette agrégation et conduit à un meilleur gonflement ce qui produit des signaux plus fins comme résultat.
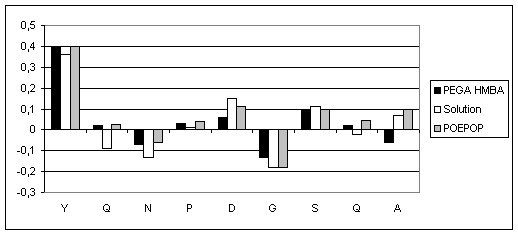
Figure 27 : Comparaison des déplacements chimiques des protons alpha du peptide sur Péga-HMBA, POEPOP et en solution par rapport à la valeur Random Coil.
Pour finaliser l’influence de la structure de la résine sur la structuration du peptide, nous avons synthétisé le peptide sur un autre type de polymère hydrogonflable.
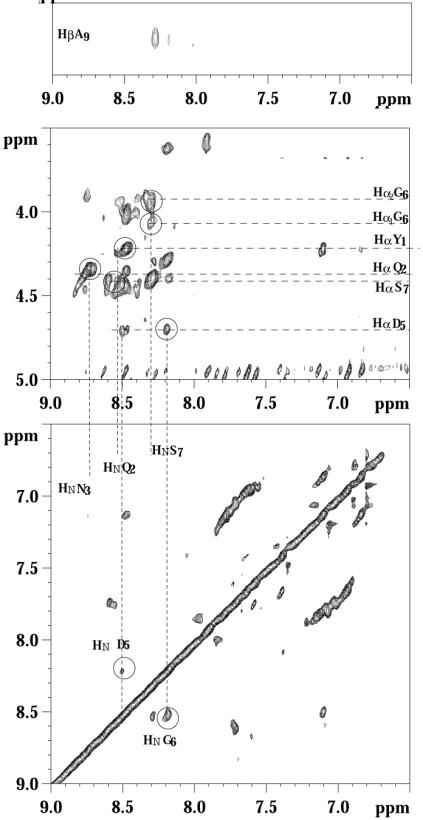
Figure 28 : Spectre NOESY zone amide du peptide YQNPDGSQA sur POEPOP 4°C, pH 3.5.
2.2.3 Formation d’un feuillet béta sur PEPSYN ?
Pour finir l’étude de l’influence du polymère sur la structuration des peptides, nous avons utilisé un troisième polymère. Cette résine (PEPSYN) est un copolymère de N, acryly sarcosine méthylester, N-N’-bisacrylyléthylène diamine et de N,N-diméthylacrylamide, dont la charge initiale est de 1 mmol/g.
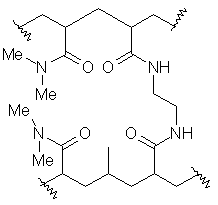
Schema 6 : Formule développée plane de la résine PEPSYN.
Comme pour les expériences précédentes, nous nous sommes placés dans les mêmes conditions que l’étude en solution, hormis pour le pH que nous avons conservé à la valeur optimale de 3,5 déterminée lors de l’étude de la structuration du peptide sur la résine Péga. Les spectres obtenus concernant le peptide sur la résine PEPSYN étaient très décevants. En effet les spectres présentaient des largeurs de ligne assez importantes et le spectre NOESY ne comportait que très peu de contacts inter-résidus (Figure 29), le peptide semblait être agrégé sur la résine.
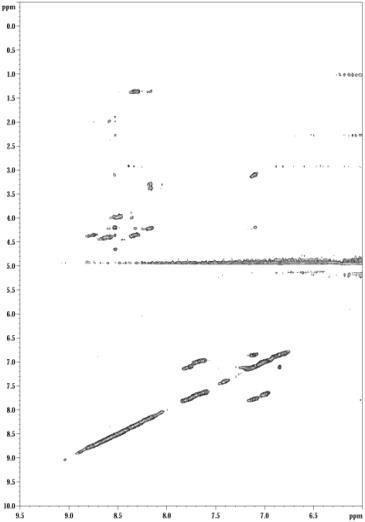
Figure 29 : Spectre NOESY zone amide du peptide béta hairpin sur PEPSYN 1 mmol/g enregistré à 4° C et à pH 3,5
Devant ce résultat surprenant, nous avons de nouveau relié la qualité médiocre des spectres à un problème de pH pour ce nouvel échantillon. Un autre échantillon du peptide a été préparé en utilisant la valeur de pH 5, déterminée pour l’étude en solution. Le changement de pH n’a pas apporté d’amélioration significative au niveau de la qualité des spectres et le spectre NOESY ne comportait pas plus de contacts NOE que dans l’échantillon précédent. Le pH étant a priori hors de cause pour l’agrégation du peptide sur la résine PEPSYN, plusieurs possibilités s’offraient à nous : soit la nature de la résine est responsable de la non structuration du peptide soit la charge très importante du polymère conduit à l’agrégation du peptide. En effet la résine PEPSYN est la résine la plus chargée que nous ayons utilisée puisque sa charge est de 1 mmol/g, alors que la résine Péga possède une charge de 0,38 mmol/g et la résine POEPOP a une charge de 0,4 mmol/g. Pour une résine conventionnelle, le diamètre d’une bille gonflée est de 200 mm, le volume de la bille est d’environ 4,2e-9 l. Il est couramment admis qu’un gramme de résine est constitué 220 000 billes.[23] Pour la résine PEPSYN, la concentration de peptide ramenée à l’échelle d’une bille est de l’ordre de 1 M, alors que pour les résines Péga et POEPOP nous obtenons une valeur de 0,4 M. La concentration importante des chaînes peptidiques sur la résine PEPSYN pourrait donc être responsable de l’agrégation du peptide sur ce polymère. En effet lorsque l’on observe les concentrations couramment utilisées pour une étude structurale détaillée par RMN, celles-ci ne dépassent que très rarement 100 mM, puisque de manière générale, une concentration plus élevée conduit inévitablement à l’agrégation de l’objet étudié. Il est vrai que dans le cas d’une étude en solution, la probabilité de rencontre des différentes chaînes peptidiques ou protéiques est beaucoup plus importante que sur la phase solide car les molécules sont libres de diffuser librement. Malgré l’absence de libre diffusion des molécules ancrées sur le support solide, une concentration trop importante de peptide conduit inévitablement à des interaction intermoléculaires conduisant à l’agrégation du peptide.
2.2.4 Conclusion
Cette étude démontre que la structuration de peptide en feuillet béta est possible sur différents supports solides, mais aussi que la charge élevée de certaines résines n’est pas compatible avec des séquences à fortes tendance à l’agrégation. Toutefois, la possibilité de décharger les résines par le biais des deux stratégies de protection orthogonale Fmoc/Boc a déjà été utilisée avec succès dans le cas du peptide poly-alanine et pourra encore être utilisée dans le cas de peptides à forte agrégation pour une étude structurale sur support solide. Il est à noter que lors de cette étude, nous avons rencontré quelques problèmes de reproductibilité d’un lot de résine à un autre. En effet, dans certains cas, les spectres 1D obtenus montraient un rapport relatif intensité du signal de PEG par rapport aux signaux dûs au peptide très favorable et pour d’autres lots de résine, ce rapport était fortement défavorable. Ce problème de reproductibilité concernant la qualité des spectres RMN est accru au cours du temps. En effet, le même lot de résine observé quelque mois après, montre un comportement complètement différent. Ce manque de reproductibilité d’un lot de résine à un autre accroît considérablement les difficultés lors de l’étude de la structuration d’un peptide sur un support solide.
2.3 Etude d’une séquence peptidique potentielle hélice alpha.
Nous nous sommes consacrés à l’étude d’un peptide formant une hélice alpha, une des majeures structures secondaires rencontrées dans les protéines. Ce type de structure secondaire a été largement étudié en solution et ces études ont conduit à de nombreux outils pouvant prédire la capacité d’une séquence à se structurer en hélice alpha.[24],[25] La séquence que nous avons étudiée est dérivée de la protéine 434 Cro d’un phage.[26] Cette protéine est constituée d’un seul domaine composé par cinq hélices. L’étude par RMN liquide haute résolution a montré que les peptides issus de cette protéine pouvaient adopter d’une façon isolée une structure en hélice alpha. Nous avons donc décidé d’étudier le peptide décrit comme ayant le plus grand pourcentage d’hélicité en solution. La séquence que nous avons étudiée est la suivante : MQTLSERLKKRRIALKY. La zone écrite en gras correspond à une zone d’hélice dans la protéine native et sera dénommée Croc dans la suite de ce chapitre. Le peptide a été synthétisé sur trois types de résines : Amino Péga, Péga HMBA, et sur PEPSYN. Nous avons choisi ces trois résines pour leur compatibilité avec un milieu aqueux et de part leur résistance au TFA lors de la déprotection des chaînes latérales du peptide.
2.3.1 Etude RMN HRMAS du peptide Cro sur résine amino Péga
Dans un premier temps, nous avons étudié la séquence entière sur le support solide pour avoir une comparaison directe avec l’étude précédemment menée en solution.. Dans un premier temps, le peptide a été synthétisé selon la stratégie Fmoc/Tbu sur une résine Amino-Péga. L’étude RMN du peptide Cro en solution, montre que le peptide adopte une conformation en hélice alpha dans les conditions suivantes : 2°C avec un pH 6.26 Le peptide Cro sur la résine Péga a donc été étudié dans les mêmes conditions. Le spectre TOCSY (figure 30) ne permet pas l’attribution complète des différents résidus constituant le peptide à cause de la trop grande répétition de certains acides aminés (voir séquence). Le spectre TOCSY montre une faible dispersion des protons alpha. La faible dispersion des protons alpha d’un peptide ou d’une protéine est souvent caractéristique d’une structuration en hélice alpha.
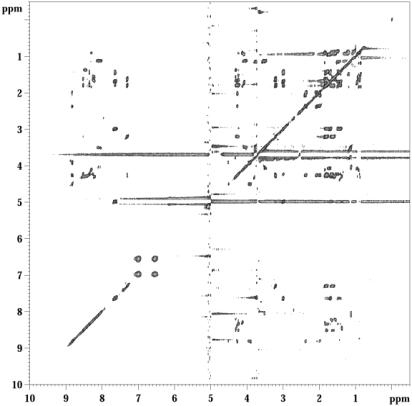
Figure 30 : Spectre TOCSY (274K) du peptide Cro long ancré sur amino Péga.
Pour attribuer totalement notre peptide et afin de lever les ambiguïtés, nous avons enregistré un spectre NOESY. Le spectre NOESY du peptide, dans les conditions décrites lors de l’étude en solution, montre de nombreux contacts NOE et en particulier des contacts NH-NH qui sont aussi une bonne indication de la structuration du peptide en hélice alpha. Toutefois ces seules indications ne sont pas suffisantes pour déterminer si oui ou non le peptide adopte une conformation en hélice alpha sur la résine. La trop grande répétition de certains acides aminés a considérablement compliqué l’attribution totale du peptide, notamment due à de nombreux recouvrements et nous avons donc décidé d’étudier seulement le domaine correspondant à l’hélice alpha dans la protéine native.
2.3.2 Etude RMN HRMAS du peptide Croc sur résine HMBA-Péga
Le peptide LSERLKKRRIAL a donc été synthétisé sur une
résine Péga-HMBA et nous avons procédé à la détermination de sa structure par
RMN HRMAS sur ce support. La séquence présente encore de nombreuses
répétitions, mais nous avons pu attribuer totalement le peptide. L’analyse des
différents contacts NOE ne nous permet pas de conclure à la structuration du
peptide en hélice alpha. Lors de l’étude en solution, Padmanabhan et col17,
ont utilisé du TFE pour favoriser la structuration en hélice alpha. Lors de
leur étude, les analyses CD du peptide en solution, donnent un pourcentage
d’hélicité de 21% pour le peptide dans l’eau et 48% pour le peptide dans 40% de
TFE. De nombreuses études ont démontré que l’utilisation de TFE favorisait la
structuration d’un peptide en hélice alpha[27].
Le TFE mime le cœur hydrophobe des protéines et par voie de conséquence,
favorise la structuration du peptide en hélice alpha. Nous avons donc procédé à
l’analyse du peptide en utilisant 40% de TFE comme solvant. Les spectres
obtenus présentent une meilleure résolution. De plus, le spectre NOESY présente
plus de contacts, en particulier les contacts NH-NH montrant que l’addition de
TFE est favorable à la structuration du peptide en hélice a (Figure 31).
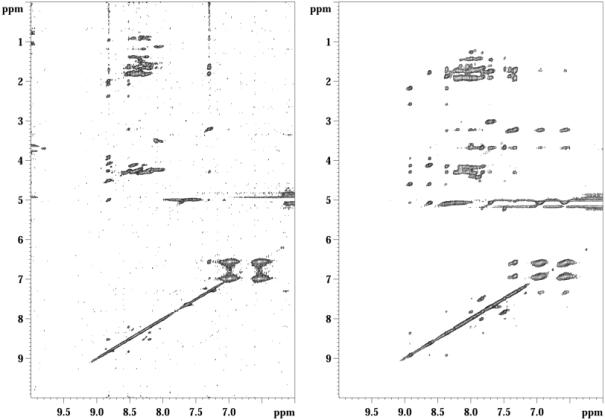
Figure 31 : comparaison des spectres NOESY zone amide de la peptidyl résine gonflée dans le tampon phosphate pH 6 (gauche) et de la peptidyl résine gonflée dans 40% de TFE.
Malgré l’amélioration de la qualité des spectres ainsi que l’augmentation du nombre de contacts NOE, il est difficile de conclure sur la structuration du peptide en hélice alpha, en particulier parce que certains contacts importants sont absents (contacts dab (i, i+3)). Nous avons vu que l’addition de TFE comme co-solvant améliorait considérablement la qualité des spectres, notamment pour les contacts NOE laissant penser que le peptide adoptait une structure en hélice alpha. Une autre bonne indication de la structuration du peptide en hélice est l'effet obtenu sur la qualité des spectres lors de l'addition d'urée. En effet, l’urée est une substance bien connue pour son effet déstabilisant sur de la structure secondaire[28]. Dans notre cas, l’utilisation d’une solution 7M d’urée réduit considérablement le nombre de contacts NOE, par rapport au spectre obtenu dans le tampon phosphate à pH 6, ce qui confirme que notre peptide adoptait une structuration en hélice alpha sur la résine (figure 32).
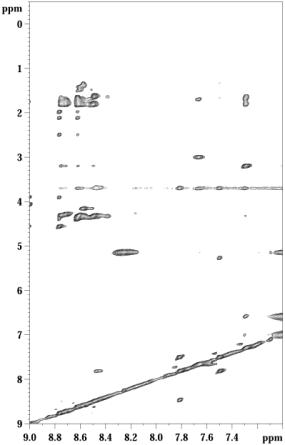
Figure 32 : Spectre NOESY zone amide de la peptidyl résine enregistré à 600 MHz dans l’urée 7M à 277K.
Devant cet échec relatif, nous avons voulu, comme dans le cas du feuillet b, étudier l’influence de la résine sur la structuration du peptide et nous avons donc utilisé un autre type de résine.
2.3.3 Etude RMN HRMAS du peptide Croc sur résine PEPSYN
La résine PEPSYN est un copolymère de N-acrylyl sarcosine méthylester, N,N’-bisacrylyéthylène diamine et de N,N-diméthylacrylamide dont la charge est de 1 mmol/g. Cette résine gonfle parfaitement dans les solutions aqueuses et est résistante au TFA lors de la déprotection des chaînes latérales. Le peptide a été synthétisé sur cette résine selon un protocole standard et nous avons procédé à l’acquisition des spectres dans les mêmes conditions que celles décrites précédemment. En comparaison avec le spectre NOESY obtenu sur la résine Péga-HMBA, le spectre NOESY était vraiment de mauvaise qualité. En effet, ce spectre ne présentait que très peu de contacts NOE, et les contacts longue distance (figure 33) sont totalement absents. Une fois de plus, le peptide semble être agrégé sur la résine, comme dans le cas du feuillet béta sur la même résine. Là encore, nous avons attribué l’agrégation du peptide à la charge élevée de la résine comme dans le cas du feuillet béta.
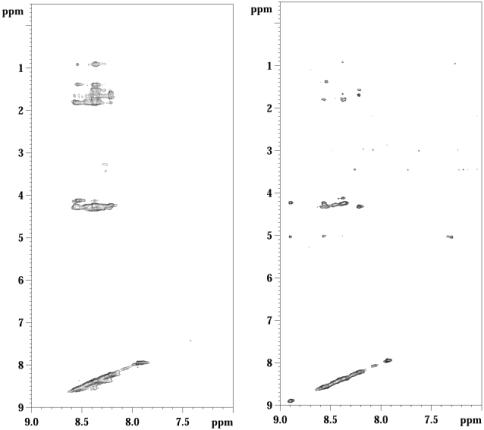
Figure 33 : Comparaison des spectres NOESY (à droite) et TOCSY (à gauche) du peptide Croc ancré sur la résine PEPSYN (1mmol/g), enregistré à 600MHz, à pH 6 et 273 K .
Ce résultat montre une fois encore que la charge important des résines peut être un obstacle à la structuration des peptides malgré une immobilisation sur la support solide. En effet, nous avons vu pour deux séquences différentes, que la charge importante sur le support solide mène vers l’agrégation.
3 Conclusion
La structuration des peptides en solution a contribué largement à l’établissement des règles de repliements en structures secondaires régulières tels que l’hélice alpha et le feuillet béta. Néanmoins le nombre de modèles adoptant une conformation en feuillet béta est nettement inférieur à ceux qui adopte une conformation en hélice alpha, surtout à cause de l’agrégation de ces premiers. Nous avons voulu évaluer si l’utilisation d’un support solide comme « chaperonne chimique » pouvait résoudre ce problème d’agrégation. La première question était celle de la compatibilité entre structure en feuillet béta et le support. En effet, alors que la présence d’une hélice alpha sur un support avait déjà été démontrée, il n’existait pas d’étude sur un feuillet béta modèle. Notre étude montre pour la première fois que l’on peut bien obtenir une structuration au moins partielle en feuillet béta. Néanmoins, lors de cette étude, plusieurs problèmes inattendus sont apparus. La haute densité des chaînes peut mener vers une agrégation temporaire, qui empêche un bon gonflement du support, ce qui se traduit immédiatement par la détérioration des spectres RMN. Un autre paramètre très important est le pH. Lorsqu’on éloigne le pH de l’étude du pI du peptide, on arrive à empêcher l’agrégation de celui-ci sur la résine. Nous pouvons comparer ce résultat avec une étude précédente en chimie : le seul ajout de DMSO au solvant de synthèse suffisait pour diminuer l’agrégation et ainsi favoriser le gonflement de la résine. Ceci conduisait à un meilleur rendement de synthèse et permettait également de récupérer des lignes fines sur les spectres RMN. La compatibilité des résines avec la structuration en feuillet béta étant prouvée, pouvons nous espérer utiliser la résine pour séparer les chaînes peptidiques ayant une tendance naturelle à agréger ? Plusieurs obstacles se présentent. D’abord, du à la flexibilité et la longueur des chaînes de PEG, il faudra probablement décharger les résines d’une telle façon qu’avant d’empêcher efficacement l’agrégation, l’étude par RMN ne sera plus possible faute de concentration. En plus, il paraît difficile d’obtenir des supports reproductibles, nous avons observé que d’un lot de résine à l’autre il était très difficile d’obtenir des résultats identiques. Pour ces raisons, il faudra plutôt se diriger vers des supports plus rigides qui sont quand même compatibles avec la synthèse peptidique et avec une solvatation en milieu aqueux. Les supports CPG utilisés en synthèse d’oligonucléotides, pourraient former un tel système, mais leur faible charge 0,1 à 100 µmole/g sera un des premiers obstacles à surmonter pour obtenir des spectres avec un rapport signal sur bruit suffisant.