Chapitre II
La RMN HRMAS et la synthčse organique en phase solide
1 Introduction
1.1 Les techniques d’analyses utilisées en phase solide
Un des premiers freins au développement de la synthčse en phase solide est le manque de techniques d’analyses rapides et précises lorsque la molécule est encore ancrée sur le support solide. La méthode la plus utilisée pour la caractérisation des molécules synthétisées en phase solide consiste ŕ séparer la molécule de son support solide pour procéder ŕ la purification de la molécule et ensuite caractériser celle-ci par les méthodes d’analyses classiques telles que la CCM, la RMN liquide haute résolution ou la spectrométrie de masse. Cette méthode est trčs efficace mais présente deux inconvénients, d’une part elle est destructrice pour le support qui ne peut pas ętre réutilisé et d’autre part, l’étape de purification nécessite beaucoup de temps. De plus la coupure chimique peut altérer la molécule synthétisée, et/ou les intermédiaires de synthčse peuvent présenter des problčmes de solubilité ou d’instabilité.
Néanmoins, lorsque le composé est encore ancré sur le support, il existe certaines méthodes qualitatives de contrôle basées sur des tests colorimétriques. Lors de la synthčse peptidique par exemple, les étapes de couplage ou de déprotections peuvent ętre contrôlées notamment par le test ŕ la ninhydrine[1],[2]. Lors de tels tests, on peut s’interroger sur leur sensibilité : jusqu'ŕ quel niveau ces techniques sont applicables. De plus, avec ces tests, on obtient un résultat global, sans aucune information au niveau moléculaire. Si l’on considčre le cas d’un acétylation irréversible d’une fonction amine lors de la synthčse d’un peptide. Le test ninhydrine aprčs l’étape de couplage donnera un résultat négatif, ce qui confirme la disparition de l’amine primaire, mais en aucun cas on ne peut savoir que l’acide aminé ancré ne sera plus réactif pour les réactions suivantes. Le problčme des tests colorimétriques et qu’ils ne sont pas généraux, ils permettent seulement de mettre en évidence quelques fonctionnalités caractéristiques.
Avant de choisir une technique d’analyse, l’expérimentateur doit s’interroger sur le but de l’analyse, la quantité de produit nécessaire et aussi la possibilité de réutilisation du support. Nous allons maintenant présenter différentes méthodes d’analyses de molécules ancrées sur un support solide avec leurs avantages et leurs inconvénients.
Différentes méthodes basées sur la spectrométrie de masse permettent une analyse de molécules ancrées sur le support solide. La spectroscopie MALDI-TOF (Matrix-Assisted Laser Desorption Ionization Time Of Flight) consiste ŕ séparer ŕ l’aide d’un laser, la molécule synthétisée du support solide. [3] Cette technique nécessite l’utilisation d’une résine particuličre car l’ancrage doit ętre photosensible. Aprčs libération du composé au sein du spectromčtre il est analysé classiquement par spectrométrie de masse. L’avantage de cette méthode est qu’elle est trčs sensible et ne nécessite que trčs peu de matériel, quelques billes de résine suffisent. Par contre, le matériel utilisé est perdu pour la suite de la synthčse. Cette technique permet une identification des molécules mais ne permet pas de quantifier les différentes espčces. La spectrométrie de masse de type SIMS (Secondary Ion Mass Spectroscopy) est une technique qui permet l’analyse de la molécule ancrée tout en conservant l’intégrité du support. Récemment Aubagnac et al[4] ont développé une méthode d’analyse non destructrice faisant appel ŕ cette technique. Elle est particuličrement utile pour les nouveaux supports solides tels que les PINS et ne nécessite pas l’emploi de « linker » photosensible. De plus cette méthode d’analyse permet d’identifier les molécules mais aussi une quantification directe sur le support[5]. Cette méthode consiste ŕ bombarder la cible par un faisceau d’ions, puis le temps de vol des ions incidents (ions secondaires) est mesuré. Ces ions sont caractéristiques de la surface de l’échantillon étudié.
L’Infra Rouge ŕ Transformée de Fourrier (IR FT) permet d’évaluer directement sur une résine le taux de déprotection lors de la synthčse peptidique ŕ condition que les groupes protecteurs soient porteurs de deutérium (Larsen, 1993 [6] ; Russel, 1996 [7]). Une autre application de la spectroscopie IR est le suivi de réaction directement sur une bille de résine[8],[9],[10]. Cette technique, bien que destructrice pour le support, permet de suivre qualitativement une transformation chimique. Cette technique comme la spectroscopie de masse n’exige que trčs peu de matériel (une seule bille doit ętre aplati avant l’acquisition afin d’obtenir de bons spectres) pour une analyse, ce qui est trčs avantageux dans le cas du suivi de longues synthčses.
Une autre technique utilisée pour l’analyse structurale de composés ancrés sur support solide est la RMN du 13C. Cette technique ne nécessite aucune modification de l’échantillon, ni de conditions particuličres pour l’analyse. La seule préparation est le gonflement de l’échantillon dans un solvant deutéré, ou protoné.[11],[12] Cette technique permet de déterminer le degré de fonctionnalisation des résines et permet également de suivre des synthčse supportées. La limite de cette technique est sa sensibilité. En effet, dű au faible rapport gyromagnétique du 13C (faible sensibilité par rapport au proton), et de sa faible abondance naturelle (1,11%), les temps d’acquisitions sont trčs longs, ce qui limite le suivi de réaction sur le support. De plus, les spectres obtenus présentent une largeur de ligne assez importante. D’autres noyaux plus sensibles tels que le 19F et le 31P ont aussi été observés. [13], [14] La vraie limite de cette technique est qu’elle ne permet pas d’observer le noyau du proton, omniprésent dans les molécules organiques et avec un comportement tout ŕ fait favorable ŕ la RMN.
Le proton n’est pas observable dans ces conditions ŕ cause de la nature hétérogčne des résines utilisées pour la synthčse en phase solide. De maničre générale toutes les résines utilisées pour la synthčse en phase solide sont organisées de la męme façon. On peut décomposer le support solide en trois zones.
Le premier domaine est la matrice solide qui constitue le corps principal du support solide. Ce support est en général une résine, c’est ŕ dire une matrice réticulée, constituée principalement de polystyrčne réticulé par du divinylbenzčne comme dans les résines Merrifield et Wang (zone 1 figure 5a), mais on trouve aussi des résines basées sur du polyéthylčne glycol (POEPOP) et ou du polyacrylamide (Pega, PEPSYN).

Le second domaine est considéré comme le bras espaceur. Ce bras espaceur est
chimiquement inerte et de longueur variable (zone 2 figure 5a). La longueur du
bras est un facteur déterminant pour la mobilité des molécules et en
conséquence, aura une grosse influence sur la qualité des spectres RMN. Enfin,
ŕ l’extrémité du bras espaceur, se trouve le groupe fonctionnel qui va servir
d’ancrage ŕ la molécule synthétisée (zone 3 figure 5a). La nature du groupement
chimique qui servira d’ancrage détermine le mode de coupure de la molécule
synthétisée du support solide.
Figure 5a : Représentation schématique d’une résine pour la synthčse en phase solide
Les différents domaines qui constituent les résines
utilisées en synthčse organique en phase solide, en font un systčme hétérogčne
avec des caractéristiques propres qui contribuent aux difficultés rencontrées
pour l’analyse de ce type de support.
Récemment une nouvelle technique RMN non destructrice a été introduite pour l’analyse directe de composés ancrés sur la phase solide. Cette technique basée sur la rotation de l’échantillon ŕ l’angle magique est la RMN MAS (Magic Angle Spinning). La RMN MAS est dérivée de la RMN du solide[15],[16]. La RMN MAS est une méthode d’analyse directe et sensible ne nécessitant aucun traitement particulier de l’échantillon si ce n’est le gonflement de la résine dans un solvant deutéré approprié. La rotation (plusieurs kHz) ŕ l’angle magique de l’échantillon permet l’observation directe des noyaux 1H, 13C, et récemment 15N, avec une résolution proche de la RMN liquide haute résolution. Notre laboratoire s’efforce d’améliorer cette technique depuis plusieurs années.
2 La RMN HRMAS une technique de choix pour la synthčse organique en phase solide
2.1 Définition de la RMN Haute Résolution ŕ l’Angle Magique
La majeure difficulté rencontrée lorsque l’on veut étudier une molécule ancrée sur un support solide par RMN est l’importance de la largeur des raies. Dans le cas d’un échantillon purement solide, la largeur de raie peut dépasser 10 kHz. Cet élargissement des raies provient notamment de l’interaction dipole-dipole (interaction dipolaire). Une autre contribution ŕ l’élargissement des raies provient des inhomogénéités de susceptibilité magnétique. Ces variations de susceptibilité proviennent de l’hétérogénéité de l’échantillon, et peuvent ętre décrites comme une interaction dipolaire entre le spin et un dipôle macroscopique.[17]
Dans le cas d’un échantillon de résine gonflée, il existe un gradient de mobilité allant de mouvements lents pour le polymčre, vers des mouvements plus rapides pour la molécule ancrée sur le « linker ». Męme si la mobilité des molécules ancrées ŕ l’extrémité du linker se rapproche des comportements rencontrés en solution, la largeur de raie reste importante et ne permet pas l’obtention de spectres exploitables. De plus, si l’on ajoute ŕ cela une inhomogénéité importante de susceptibilité magnétique due ŕ l’importante hétérogénéité de l’échantillon cette inhomogénéité contribue aussi ŕ l’élargissement. En effet lorsque la résine est gonflée, elle n’est plus solide pur et comporte différentes zones, certaines contenant du solvant, d’autres étant constituées de domaines non solubles ŕ mobilité réduite, ou encore des domaines de grande mobilité. Toutes ces zones vont créer ŕ l’intérieur de l’échantillon des grandes différences de susceptibilité magnétique, qui ŕ leur tour vont contribuer ŕ l’élargissement de raies.
La RMN HRMAS appliquée ŕ la synthčse en phase solide repose sur deux conditions. Contrairement ŕ la RMN du solide, l’échantillon doit ętre gonflé dans un solvant approprié. Le bon gonflement du polymčre garantit une mobilité importante des molécules ancrées grâce ŕ leur solvatation adéquate et réduit au moins partiellement les interactions dipolaires qui sont propres aux solides. Nous avons mentionné que dans le cas de la RMN gel, le simple gonflement de la résine permet l’obtention de spectres 13C mais ne permet pas l’obtention de spectres 1H. Du fait du rapport gyromagnétique élevé du proton, l’interaction dipolaire et les effets de susceptibilité magnétique sont exacerbés et dans ce cas le gonflement de la résine n’est pas suffisant pour moyenner ces interactions. La deuxičme condition, est que l’enregistrement des spectres doit ętre effectué en utilisant la technique de rotation de l’échantillon ŕ l’angle magique. Tandis que le gonflement de la résine permet une solvatation des molécules et leur confčre ainsi une mobilité accrue par rapport ŕ la résine sčche, la rotation rapide de l’échantillon ŕ l’angle magique permet de moyenner les différences de susceptibilité magnétique, ainsi que l’effet dipolaire résiduel, ce qui permet d’obtenir des spectres résolus en 1H, 13C pour la molécule ancrée sur le support.
En combinant le gonflement de la résine et le « magic angle spinning », le comportement des signaux RMN des molécules ancrées sur le support solide tend ŕ se rapprocher des composés rencontrés en RMN liquide haute résolution (figure 5b). Toutes les séquences impulsionnelles utilisées sont celles empruntées ŕ la RMN du liquide.
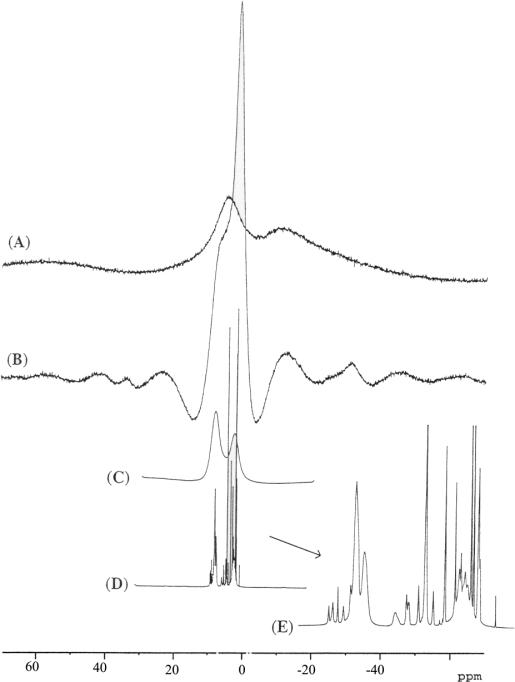
Figure5b : Spectres 1D 1H
de la peptidyl résine Wang-Ile-Val-Ser(OtBu)-Gly-Arg(Pmc)-Ala-H.
(A) Spectre statique de la peptidyl résine sčche. (B) spectre sous MAS (wr = 5kHz) de la peptidyl résine sčche. (C) Spectre statique de la peptidyl résine gonflée dans le DMF-d7. (D) et (E) spectre sous MAS (wr = 4kHz) de la peptidyl résine gonflée dans le DMF-d7.
2.1.1 Interaction dipolaire
Comme nous venons de le voir, l’interaction dipolaire est en grande partie responsable de l’élargissement important des signaux obtenus pour un spectre d’une résine gonflée dans un solvant. Si l’on représente graphiquement le champ dipolaire (figure 6) pour une paire isolée de spins, on constate qu’il existe une orientation pour laquelle le champ dipolaire, sans ętre nul, est perpendiculaire au champ magnétique statique B0.
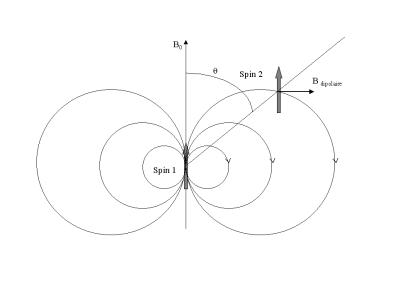
Figure 6 : Représentation graphique du champ dipolaire d’un spin 1 orienté selon B0. Ce champ n’a plus de composante selon z quand q est de 54°,7 solution de 3 cos2(q)-1=0.
L’interaction dipolaire, qui est le produit scalaire du moment magnétique du spin avec le champ dipolaire est alors réduite ŕ zéro. L’orientation pour laquelle l’interaction dipolaire est réduite ŕ zéro correspond ŕ un angle de 54°,7 par rapport au champ statique B0, cet angle est appelé l’Angle Magique. Le seul fait de basculer l’échantillon ŕ l’angle magique n’est pas suffisant pour obtenir des spectres de qualité satisfaisante. Si l’on considčre un échantillon solide, il est évident que toutes les paires de spin ne peuvent pas toutes ętres orientées selon un axe orienté ŕ 54,7° par rapport ŕ B0. La rotation rapide de l’échantillon selon un axe orienté ŕ 54,7° par rapport ŕ B0 va par contre imposer une orientation moyenne pour toutes les paires de spin qui coďncident avec l’axe du cône de rotation. Ainsi l’élargissement résiduel des raies (dű aux inhomogénéités du champ magnétique) est inférieur ŕ 10 kHz, et est moyenné par la rotation rapide de l’échantillon ŕ une vitesse supérieure ŕ l’interaction que l’on veut moyenner. Une rotation supérieure ŕ 1kHz permet d’obtenir des signaux suffisamment fins pour les molécules ancrées sur le support alors que les raies dues au polymčre conservent une largeur importante de part leur mobilité restreinte. Cette différence de mobilité entre les signaux du polymčre de la molécule ancrée sur le support et le solvant, peut ętre mise ŕ profit lors des expériences HRMAS.
2.1.2 RMN HRMAS et le filtre de diffusion
La séquence d’impulsion du filtre de diffusion est une séquence d’impulsion permettant de s’affranchir des signaux des molécules pouvant diffuser librement dans la matrice solide. Les molécules pouvant diffuser librement sont les molécules de solvants ou encore les excčs de réactifs dans le cas de transformations incomplčtes. Le séchage des résines pour le regonflement dans un solvant deutéré est une opération qui nécessite beaucoup de temps et il est parfois utile de pouvoir caractériser une molécule rapidement comme dans le cas d’intermédiaires de synthčses sensibles, tels que les énolates.[18]
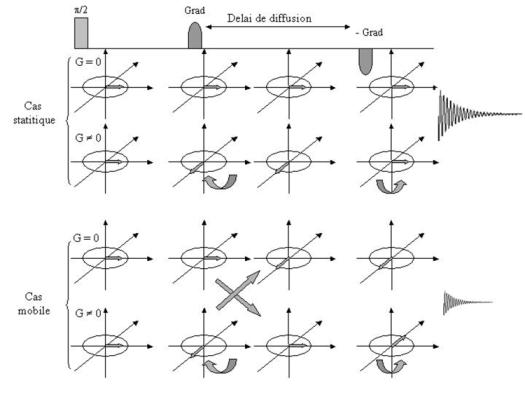
Figure 7 : Séquence d’impulsion simplifiée du filtre diffusion et son influence sur le vecteur aimantation.
Comme nous l’avons vu précédemment, dans les échantillons de résines solvatées, il existe un gradient de mobilité entre les molécules ancrées sur la résine et les molécules libres en solutions. Dans certains cas ce gradient de mobilité peut ętre mis ŕ profit. En effet les molécules greffées sur le support solide, bien que possédant une mobilité importante due ŕ la libre rotation autour des liaisons, ont perdu la faculté de diffuser librement (mobilité translationnelle perdue de part l’ancrage sur la matrice solide) ŕ l’intérieur de l’échantillon. Les molécules constituant le solvant, peuvent par contre diffuser librement dans tout l’échantillon. Cette différence de mobilité translationnelle a été mise ŕ profit au laboratoire pour supprimer les signaux dus au solvant[19]. La séquence d’impulsion du filtre diffusion repose sur l’utilisation de gradients qui sont utilisés pour défocaliser l’aimantation (Figure 7).
Dans la figure précédente, on constate que pour les molécules ancrées (cas statique figure 7), l’utilisation des gradients ne conduit pas ŕ une diminution du signal car lors de l’application du deuxičme gradient, la molécule étant toujours ŕ la męme place, l’aimantation est re-focalisée. Par contre pour les molécules ŕ forte mobilité, (cas mobile figure 7), l’application du deuxičme gradient ne re-focalise pas l’aimantation car pendant le délai de diffusion, les molécules se sont déplacées et ne ressentent donc pas le deuxičme gradient avec la męme intensité que le premier. L’aimantation n’étant pas re-focalisée, cela conduit ŕ une perte de signal. Lorsque nous utilisons le filtre de diffusion au laboratoire, nous utilisons une puissance de gradient de 35 G/cm pendant une durée de 5 ms et un délai de diffusion de 30 ms. 35 G/cm représente 150000 Hz/cm. L’évolution de l’aimantation d’un spin2 sur une molécule « libre » relatif ŕ celle de d’un spin1 au centre de l’échantillon, sera différente par Dw.Dt ou Dw=2p.15000Hz. Le vecteur 2 tourne de 75 tours dans le référentiel ou le vecteur 1 est statique. Inversement nous pouvons calculer la distance par rapport au centre de l’échantillon qui provoquera une simple inversion (évolution de l’aimantation p) de l’aimantation, le calcul nous donne une distance de 6µm. Si le délai de diffusion permet aux molécules mobiles de se déplacer sur une distance de 6µm au moins, l’orientation de leur aimantation ne sera plus corrélée avec celle de la molécule au centre et nous perdrons du signal. Le coefficient de diffusion de l’eau liquide est de 2e-9 m2/s. Pendant les 30ms du délai de diffusion, une molécule d’eau se déplace de10µm dans le sens de l’axe du gradient. Ceci signifie que durant le délai de 30ms, les molécules mobiles auront suffisamment de temps pour changer de place et ne plus voir leur aimantation refocalisée. Le filtre diffusion en RMN HRMAS permet donc de s’affranchir des signaux dus aux solvants, ce qui se révélera trčs utile pour les études présentées dans la suite de ce chapitre.
La technique RMN HRMAS permet donc d’avoir un accčs direct ŕ l’identification complčte des molécules attachées au support solide en cours de synthčse. Il est aussi possible de déterminer les problčmes structuraux qui peuvent ętre ŕ l’origine de couplages difficiles[20]. Enfin, une analyse conformationnelle de la molécule basée sur l’analyse des contacts NOE (Würtrich, 1986)[21] et sur la déviation des déplacements chimiques 1H, 13C et 15N ( Wishart, 1992, 1994, 1995, Fruchart 2000)[22],[23],[24],[25] peut également ętre effectuée. L’étude conformationnelle sur support solide peut avoir un avantage considérable sur la RMN haute résolution liquide pour les séquences peptidiques ayant une forte tendance ŕ l’agrégation.
2.2 Applications de la RMN HRMAS ŕ la synthčse organique en phase solide
Récemment la technique RMN HRMAS combinée au gonflement de la matrice solide, s’est révélée ętre la méthode d’analyse de choix pour caractériser les composés organiques sur support solide. Au laboratoire, nous avons déjŕ montré que la technique permettait de suivre une transformation chimique,[26] l’utilisation du filtre de diffusion a permis d’effectuer le suivi en solvant protoné,17 et récemment nous avons montré que cette technique permettait la quantification des espčces impliquées dans une transformation chimique25. Contrairement ŕ la spectroscopie de Masse ou ŕ L’IR, la RMN HRMAS ne permet pas de travailler ŕ haut débit notamment ŕ cause du temps de préparation de l’échantillon. Comme nous l’avons vu dans le chapitre d’introduction, la synthčse en phase solide est un outil intéressant pour la synthčse de molécules complexes mais aussi pour l’obtention des banques de molécules de façon combinatoire. Comme nous voulons mettre au point une technique d’analyse sur support solide, il faut répondre ŕ deux questions : comment suivre une réaction de façon quantitative et comment détecter rapidement les impuretés éventuelles ? Les récents développements de la RMN HRMAS au laboratoire ont permis de répondre ŕ la premičre question concernant le suivi quantitatif d’une réaction, mais le problčme de la détection rapide d’impuretés reste entier. Dans les paragraphes suivants nous verrons comment la RMN HRMAS peut apporter une solution ŕ ce problčme et nous nous intéresserons également ŕ l’analyse de supports macroscopiques qui connaissent une utilisation croissante.
2.2.1 Suivi d’une condensation de Wittig-Horner Emmons sur un support macroscopique.
Notre groupe a déjŕ appliqué la RMN HRMAS pour le suivi de réaction sur les résines classiquement utilisées en SOPS. [27] La manipulation de ces résines pour le suivi de réaction n’est pas toujours évidente, en particulier dans le cas de réactions rapides. Pour suivre une réaction sur les résines, il faut prélever des aliquots ŕ intervalles réguliers pour les introduire dans le rotor HRMAS afin d’enregistrer les spectres. Le remplissage du rotor n’est pas toujours aisé et l’acquisition des spectres nécessite du temps. Pendant l’acquisition des spectres sur l’échantillon prélevé, la réaction se poursuit. Les prélčvements successifs de quelques milligrammes de résine peuvent déséquilibrer la réaction, de plus les aliquots ne représentent pas la totalité du milieu réactionnel et on peut faire face ŕ des problčmes de distribution statistique.
Comme nous l’avons vu dans le chapitre d’introduction, le développement de la chimie combinatoire a contribué au développement de nouveaux supports solides, tels que les PINS ou les lanternes développées par Mimotopes. Ces nouveaux supports sont trčs utilisés en particulier dans le cas de la stratégie « one bead one compound ». De plus ces nouveaux supports ont trouvé un intéręt particulier dans le cadre des synthčses automatisées. Compte-tenu des difficultés déjŕ importantes de suivi de réaction sur les supports solides classiquement utilisés en phase solide, il est important de voir si ces nouveaux supports permettent une analyse directe des molécules ancrées sur leur surface. L’analyse directe de molécules sur ce type de support a déjŕ été réalisée par d’autres techniques telles que la spectrométrie de masse4 ou encore l’IR[28]. Shapiro et col[29]ont montré qu’il était possible d’obtenir des spectres 1D et 2D par RMN HRMAS complčtement exploitables sur ce type de support mais ceci ŕ l’aide d’une sonde 7 mm et de plus aprčs échange du solvant avec un solvant deutéré. Récemment Gerritz et al[30] ont étudié des copeaux de lanternes dans une sonde 4 mm. Bien que la sonde 4 mm soit un équipement plus conventionnel que la sonde 7 mm, le fait de raser les lanternes est une méthode destructrice et se rapproche du cas du suivi de réactions sur les résines conventionnelles.
Il est vrai que les dimensions actuelles des supports macroscopiques tels que les lanternes ne sont pas compatibles avec une étude directe de ce type de support dans un rotor 4 mm (figure 8).

Figure 8 : De gauche ŕ droite : Capuchon de rotor 4mm, rotor HRMAS 4 mm, lanterne synphase ( 35 mmoles), microbeads (1400 nmoles).
Compte-tenu de ce problčme de dimension, nous avons coupé une lanterne en quatre dans le sens de la hauteur. Bien que cette coupure du support ne soit pas satisfaisante, elle nous permet de conserver la nature macroscopique du support. Le quart de lanterne ainsi obtenu a été directement utilisé pour effectuer le suivi RMN HRMAS de la réaction de Wittig-Horner Emmons (Schéma 1), afin de comparer nos résultats avec ceux obtenus lors du suivi de la męme réaction sur une résine standard.
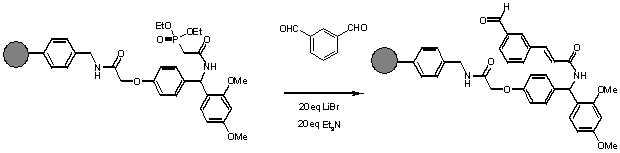
Schéma 1: Condensation de Wittig-Horner Emmons réalisé sur une lanterne SP-PS-D-RAM (fonctionnalisée par une linker Rink 35 mmoles).
Comme le montre le schéma réactionnel précédent, le térephtalaldéhyde a été ajouté sur le phosphodiester ancré sur la résine et nous avons procédé au suivi RMN afin de mettre en évidence l’apparition du signal de l’aldéhyde qui n’est pas impliqué dans la condensation.
Dans un premier temps, nous avons enregistré des spectres 1D dans le DMF deutéré afin de déterminer les conditions optimum pour effectuer le suivi de la réaction. Les spectres obtenus donnent des résultats satisfaisants mais afin d’optimiser les paramčtres du filtre diffusion, nous avons aussi enregistré des spectres du quart de lanternes dans les conditions du filtre diffusion. En accord avec les résultats précédemment décrits25, un délai de diffusion de 28 ms ainsi qu’une puissance de gradients de 35 G/cm, donnent des résultats satisfaisants. Non seulement le filtre de diffusion permet une suppression efficace des signaux du solvant, mais il permet aussi d’atténuer fortement l’intensité des signaux de la matrice polymčre. Cette intensité habituellement atténuée par l’utilisation de la séquence CPMG[31],[32], est ici largement atténuée par la relaxation T2, lors de l’application des gradients (figure 9). La séquence CPMG consiste en une suite d’impulsions 180° qui ont pour but de refocaliser l’aimantation. Durant l’application de ces impulsions, l’aimantation se trouve dans le plan xy et en conséquence, on aura une perte d’aimantation par relaxation T2 pour les molécules ŕ faibles mobilités. Dans le cas du filtre diffusion, la męme perte d’aimantation par relaxation T2 se produit lors de l’application des gradients. Aprčs la phase d’optimisation en solvant deutéré, nous avons de nouveau analysé l’échantillon en solvant protoné. Lŕ encore, le filtre diffusion permet de supprimer efficacement les signaux du solvant mais aussi atténue l’intensité de la ligne due ŕ la matrice polymčre.
Pour effectuer le suivi de la réaction, le quart de lanterne a été introduit dans une solution contenant l’aldéhyde, puis la base et le catalyseur ont été rajoutés. Avant l’introduction de la lanterne dans le spectromčtre le quart de lanterne a été abondamment lavé avec du DMF afin de stopper la réaction. Un lavage abondant de la lanterne est trčs important pour éliminer totalement l’aldéhyde n’ayant pas réagi. En effet, lors du premier essai, le spectre que nous avons obtenu présentait un signal d’aldéhyde double. Un nouveau lavage de lanterne nous a permis d’éliminer le signal provenant de la fraction soluble du térephtalaldéhyde (figure 10).
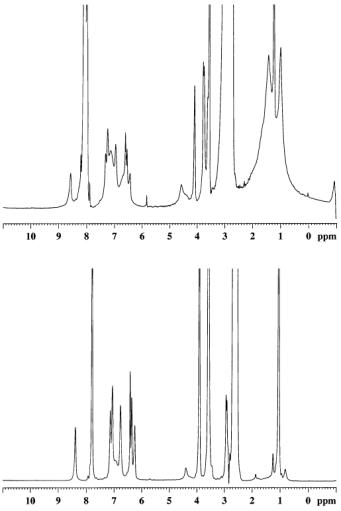
Figure 9 : Comparaison des spectres 1D d’un quart de lanterne SP-PS-D-RAM, avant réaction. L’acquisition des spectres a été effectuée sur un spectromčtre Bruker DMX 600. En haut le spectre présente des signaux intenses ŕ 1,5 ppm provenant de la matrice polymčre. Ces signaux sont largement atténués dans le cas du filtre diffusion (bas).
Comme la lanterne représente la totalité du milieu réactionnel le fait de la laver efficacement conduit ŕ un arręt de la réaction, ce qui permet de supprimer les contraintes de temps pour la préparation de l’échantillon RMN. Dans l’introduction de ce chapitre nous avons discuté de différentes techniques d’analyses et nous avons notamment abordé la notion de destructeur. Le terme de destructeur peut ętre considéré de deux façons : Soit la technique ne nécessite que trčs peu de matériel mais le support analysé est perdu pour la suite de la synthčse, soit l’analyse nécessite quelques milligrammes de résines mais le support est réutilisable. Le problčme rencontré avec les résines est que les transferts du milieu réactionnel vers le rotor HRMAS ainsi que les transferts inverses, conduisent inévitablement ŕ des pertes. Lors d’une synthčse multi-étapes longue, les transferts engendrent trop de pertes ce qui n’est pas satisfaisant. La nature macroscopique des lanternes ne conduit ŕ aucune perte de matériel ce qui est trčs avantageux au regard des résines conventionnelles. Afin d’obtenir un bon rapport signal sur bruit en vue d’une intégration future, nous avons enregistré chaque spectre avec 256 scans. Aprčs chaque spectre, le quart de lanterne était de nouveau introduit dans le milieu réactionnel. L’avancement de la réaction a été mis en évidence en mesurant l’intégrale du pic de l’aldéhyde au cours du temps. Pour l’intégration du pic de l’aldéhyde, nous n’avons pas besoin de références externes contrairement ŕ l’étude sur la résine car dans le cas présent, la totalité de l’échantillon se trouve dans le rotor (la quantité de produit observée reste constante) (figure 11). La courbe obtenue n’est pas quantitative mais révčle seulement de l’avancement relatif de la réaction. Un avancement quantitatif nécessite comme dans le cas de l’étude sur la résine, l’emploi d’une référence interne qui sert ŕ étalonner la réaction.
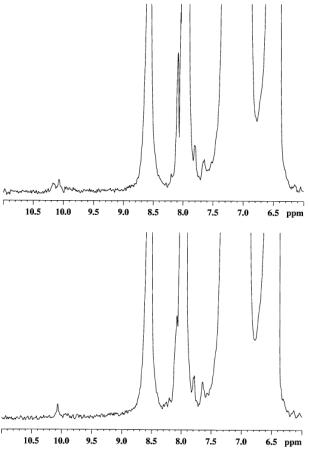
Figure 10 : Spectres 1D avec filtre diffusion, en haut lanterne aprčs un simple lavage présentant un signal d’aldéhyde double dű ŕ la fraction d’aldéhyde n’ayant pas réagi. Le spectre du bas ne présente plus cette résonance aprčs un triple lavage de la lanterne par du DMF.
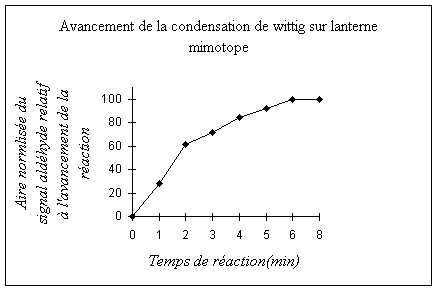
Figure 11 : Avancement de la réaction provenant de l’intégrale du signal de l’aldéhyde
La forme de la courbe obtenue est en bon accord avec celle obtenue sur la résine, par contre dans le cas de la lanterne, la cinétique de réaction est plus rapide. En effet dans le cas de la résine la réaction était complčte aprčs 7h, dans notre cas la réaction est complčte aprčs 10 min. Cette différence concernant la cinétique de réaction peut trouver une explication dans la nature du support utilisé. En effet, la résine utilisée dans l’étude précédente est basée sur une matrice polystyrčne qui par sa réticulation peut limiter la bonne diffusion des réactifs ŕ l’intérieur de la résine, alors que les lanternes sont normalement fonctionnalisées en surface. Une fonctionalisation uniquement basée sur une charge de surface ne semble pourtant pas ętre compatible avec une charge aussi élevée. Une étude de la structure des lanternes a été effectuée par spectroscopie RAMAN. Cette étude montre que la couche fonctionnelle a une épaisseur de 50 mm. Cette épaisseur de 50 mm correspond au diamčtre d’une bille dans les résines conventionnelles. Le polymčre constituant la couche fonctionnelle est un polymčre qui n’est peu ou pas réticulé. Cette absence de réticulation permet une meilleure diffusion des réactifs dans la matrice polymčre, ce qui peut expliquer la cinétique rapide constatée sur les lanternes par rapport aux résines conventionnelles qui elles sont réticulées.
Dans la premičre partie de notre étude nous avons mis en évidence l’apparition d’un signal d’aldéhyde. Cette résonance, qui a un déplacement chimique trčs caractéristique, permet donc une identification plus aisée. Comme toutes les synthčses organiques en phase solide ne sont pas toujours basées sur des fonctions ayant un déplacement chimique aussi caractéristique, il est important de déterminer si la spectroscopie ŕ deux dimensions est possible sur ce type de support macroscopique. Afin d’identifier toutes les résonances présentes sur la lanterne, nous avons enregistré un spectre TOCSY et un HSQC sur le quart de lanterne aprčs réaction (Figure 12). Les spectres obtenus montrent que l’identification de molécules complexes peut parfaitement ętre effectuée sur ce type de support montrant une nouvelle fois l’importance de l’apport de la RMN HRMAS pour la chimie organique en phase solide.

Figure 12 : Spectres 2D sur l’échantillon aprčs condensation de Wittig-Horner. Le spectre TOCSY a été enregistré en DMF protoné. Le spectre HSQC 1H-13C a été enregistré en DMF-d7. Le pic de corrélation de l’aldéhyde est visible dans la zone marquée par une astérisque.
2.2.2 Détection d’impuretés en chimie organique en phase solide
2.2.2.1 Détection d’impuretés sur un support macroscopique
Avec le développement de la chimie combinatoire, le problčme de la détection d’impuretés est devenu un enjeu crucial. Lors de la mise au point d’un type de réaction sur support solide, il est impossible de tester les conditions de la réaction pour toutes les molécules qui vont ętre utilisées. L’optimisation est réalisée seulement sur quelques composés, en considérant que les conditions applicables ŕ ces composés seront applicables ŕ toutes les molécules de la męme famille. La détection précoce d’impuretés dans le cadre de ces réactions tests est donc un enjeu d’importance pour éviter par la suite la présence de molécules non identifiées, potentiellement actives.
Aprčs avoir montré que le suivi d’une réaction męme rapide était possible sur un support macroscopique, nous nous sommes donc intéressés ŕ la détection d’impuretés sur ce type de support. Pour ce faire, nous avons donc couplé simultanément deux types d’aldéhydes sur la męme lanterne. Afin d’éviter tous les problčmes liés ŕ une différence de réactivité entre les deux espčces, nous avons couplé 1 équivalent d’un mélange contenant 80% de 3-cyclohexen-1-aldéhyde et 20% de térephtalaldéhyde, pendant 10 min, cette durée correspondant au temps de réaction déterminé précédemment. Pour assurer un couplage total, 1 équivalent du mélange a de nouveau été engagé pour un autre couplage. Le spectre 1D permet la différenciation des deux espčces, de plus l’intégration des différents pics conduit ŕ une valeur de 81 % pour le produit de ŕ la condensation du 3-cyclohexen-1-aldéhyde et 19 % pour le produit de condensation du térephtalaldéhyde (figure 13).

Figure 13 : Spectres 1D (temps de relaxation 3s) du quart de lanterne aprčs couplage simultané des deux espčces. L’intégrale entre 5 et 6 ppm représente l’espčce majoritaire, alors que le signal d’aldéhyde ŕ 10 ppm représente l’espčce minoritaire
Ces valeurs sont en bon accord avec le pourcentage théorique initialement introduit, montrant que la RMN HRMAS est une technique trčs intéressante pour l’analyse de réaction sur des supports macroscopiques.
2.2.2.2 Détection quantitative d’impuretés sur résine
Dans le cadre d’une collaboration avec les Profs. Aubagnac et Martinez du LAPP de Montpellier (Laboratoire des Aminoacides, Peptides et Protéines), nous avons voulu tester les capacités de la RMN HRMAS pour la détection d’impuretés sur les résines couramment utilisées en synthčse organique en phase solide. En utilisant des quantités différentes de Glutamate et Pyroglutamate, Enjalbal et al 15 ont créé un systčme artificiel contenant un pourcentage contrôlé d’impureté. L’étude de cette transformation n’est pas anodine car ce type de modification de l’acide glutamique peut se produire lors de la coupure du peptide en milieu acide. Ils ont ensuite procédé ŕ l’analyse de leur systčme par spectroscopie de masse (SIMS) et ils ont comparé les résultats obtenus par cette technique avec les pourcentages déterminés par HPLC aprčs séparation des molécules du support solide.5 Malgré la bonne corrélation entre les résultats de l’analyse SIMS et ceux obtenus par HPLC, confirmant la spectroscopie de masse comme une technique sensible pour la détection d’impuretés, la détection d’impuretés par la méthode SIMS pose certains problčmes notamment dus ŕ des problčmes d’ionisation inhomogčne ou encore dus ŕ une distribution non homogčne des molécules sur les différentes billes de résine, puisque lors de ces analyses, seules les molécules ancrées en surface sont accessibles.
Nous avons donc étudié le męme systčme pour connaître les possibilités et limites de la RMN HRMAS vis ŕ vis de la détection et quantification d’impuretés.
2.2.2.3 Identification des différentes espčces
Avant de procéder ŕ une étape de quantification, il est primordial d’identifier les espčces en présence pour voir si le systčme est compatible avec une quantification ultérieure. Pour procéder ŕ l’identification, nous avons enregistré les spectres TOCSY de trois échantillons, tout d’abord les deux échantillons contenant 100% de chaque espčce ainsi que l’échantillon contenant un mélange équimolaire de Glutamate et de Pyroglutamate (figure 14).
|
a |
|
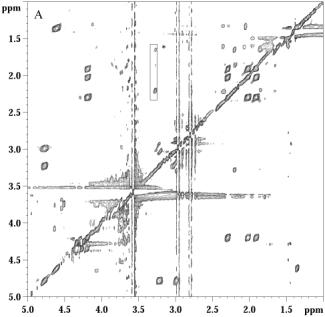
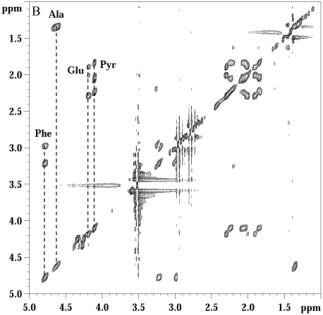
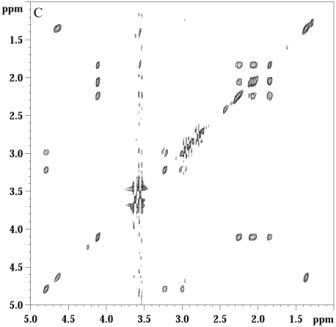
Figure 14 A) Spectre HRMAS TOCSY partie aliphatique sur 3 mg de l’échantillon contenant 100% de Glutamate. B) Spectre HRMAS TOCSY partie aliphatique sur l’échantillon, contenant un mélange équimolaire de Glu/Pyr. C) Spectre HRMAS TOCSY partie aliphatique sur 3mg de résine contenant 100% de Pyr.
Ces spectres nous ont permis d’identifier toutes les résonances provenant de chaque acide aminé, de plus nous pouvons constater que les protons alpha du Glutamate et du Pyroglutamate ne résonnent pas ŕ la męme fréquence. La présence de résonances distinctes est un point crucial pour pouvoir procéder ŕ une étape de quantification. En effet pour procéder ŕ la quantification des différentes espčces, nous devons intégrer les signaux correspondants. Dans le cas oů deux résonances sont trčs proches, l’intégration des différents signaux peut ętre délicate et conduire ŕ des résultats faussés. La nécessité d’obtenir des signaux parfaitement séparés met en évidence le besoin de travailler avec des hauts champs. Si l’on considčre de nouveau les deux résonances voisines, elles ne seront pas suffisamment séparées sur un 300 MHz, mais elles le seront sur un 600 MHz.
2.2.2.4 Analyse quantitative des différentes résines
Pour procéder ŕ la quantification des espčces, nous avons utilisé les spectres proton ŕ une dimension des différents échantillons (figure 15). La proximité des protons CH du groupement protecteur Fmoc avec le proton alpha du Glutamate n’étant pas favorable, nous avons utilisé le proton alpha du Pyroglutamate pour effectuer la quantification des différentes espčces. La quantification peut se réaliser trčs facilement par intégration du signal correspondant au proton a du Pyroglutamate par rapport ŕ une référence. Il faut se remémorer que chaque échantillon contient un pourcentage variable de Glu et Pyr, mais contient par contre une quantité constante de Phe et de Ala (représentant dans tous les cas 100% des chaînes greffées sur la résine). Les protons alpha de l’alanine ou de la phénylalanine peuvent alors servir de référence pour les intégrations. Nous avons donc utilisé ce principe pour notre étude et nous avons comparé nos résultats avec les résultats obtenus par spectroscopie de masse et aussi avec les valeurs de référence obtenues par HPLC aprčs coupure (Tableau1).
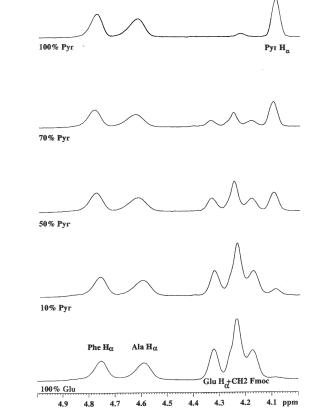
Figure 15 Spectre 1D des différentes résines gonflées dans le DMF-d7. Spectres enregistrés sur un spectromčtre BRUKER DMX 600MHz avec 64 scans et un délai de 3s.
Aprčs traitement des différents spectres ŕ une dimension, nous avons obtenu des résultats proches des valeurs obtenues par HPLC aprčs coupure, démontrant que la RMN HRMAS est un outil performant aussi bien pour l’identification que la quantification de molécules ancrées sur un support solide.
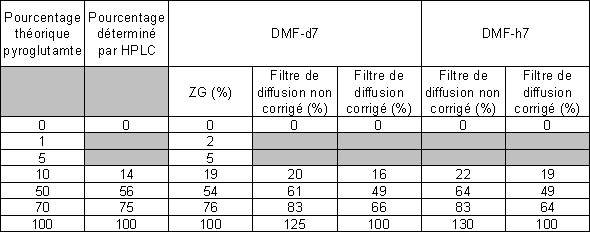
Tableau 1 : Tableau récapitulatif des pourcentages de pyroglutamate déterminés par les différentes techniques.
Aprčs ces résultats concluants, nous avons voulu déterminer le seuil de sensibilité de notre technique. Pour ce faire, nous avons réalisé un échantillon contenant 1% de pyroglutamate. Aprčs coupure, nous n’avons pas pu mettre en évidence la présence de pyroglutamate par HPLC. Par contre l’acquisition d’un spectre ŕ 1D dans les męmes conditions que précédemment, nous ŕ conduit ŕ une valeur de 2%. Pour déterminer les limites de la technique, nous avons effectué l’acquisition d’un spectre TOCSY (acquisition sur une nuit 72 scans) sur 10mg de la résine contenant 1% de pyroglutamate. A notre surprise, le spectre ne montrait pas seulement les corrélations dues au pyroglutamate, mais aussi un systčme de connectivité trčs proche de celui du glutamate (figure 16). Compte tenu du fait que l’intensité de ce systčme de spin est de l’ordre de 1%, nous avons attribué ces résonances ŕ la fraction de Glu se trouvant ŕ la surface des billes de résine. De plus, les męmes résonances mineures sont visibles pour l’échantillon contenant une quantité équivalente de Glu et de Pyrglu , ainsi que pour les résonances des Ha de l’alanine et de la phénylalanine. Il est couramment admis[33] que seulement 1% de la charge totale se trouve ŕ la surface des résines ce qui est en bon accord avec la valeur que nous avons déterminée par intégration du pic de corrélation.
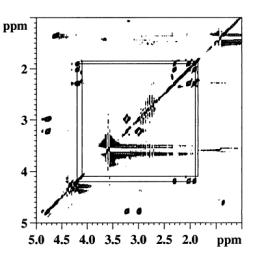
Figure 16 : Spectre TOCSY zone aliphatique sur 10 mg de résine contenant 1% de pyroglutamate.
La différence de déplacement chimique observée pour les molécules se trouvant en surface peut s’expliquer par une différence de susceptibilité magnétique. Les molécules se trouvant proches du cśur de la résine se trouvent dans un environnement fortement aromatique dű ŕ la présence du polymčre, alors que les molécules de surfaces sont seulement en contact avec le solvant qui n’a pas un caractčre aromatique. Afin de vérifier notre hypothčse, nous avons enregistré un spectre TOCSY sur la męme résine mais en utilisant du benzčne. Le spectre résultant ne présente pas ces résonances supplémentaires, ce qui confirmait l’hypothčse de l’observation de molécules de surface. Dans cette expérience, le benzčne a été utilisé pour mimer l’environnement aromatique du cśur de la résine et pour diminuer les inhomogénéités de champ magnétique. Un ré-examen des différents spectres 2D nous a permis de mettre en évidence une résonance inattendue dans le spectre de l’échantillon contenant 100% de glutamate (figure 14A encadré). Nous avons attribué ce systčme de spin ŕ une fraction de glutamate ayant été partiellement deprotégé. Pour contrôler notre hypothčse, une fraction de la résine a été déprotégée et le spectre obtenu présentait les męmes résonances. Cette déprotection partielle du glutamate est due ŕ un stockage prolongé de la résine dans du DMF. La mise en évidence de cette déprotection illustre bien l’efficacité de la RMN HRMAS pour l’identification d’impuretés, mais aussi la sensibilité de la technique. On peut se demander si dans ce cas, les tests colorimétriques comme le test ninhydrine aurait pu mettre en évidence cette déprotection de la résine. Ces tests colorimétriques sont révélateurs de certaines fonctionnalités (ils ne sont donc pas universels) et peuvent dans certains cas conduire ŕ des faux positifs ou ŕ des faux négatifs. La sensibilité de ces tests vis ŕ vis de la RMN HRMAS est ŕ démontrer car ils conduisent ŕ une réponse binaire alors que la RMN permet une identification complčte des molécules ancrées. Toutefois les tests colorimétriques sont rapides et ne nécessitent que trčs peu de résines contrairement ŕ la RMN.
Au début de notre étude, nous avons utilisé un solvant deutéré qui nécessite donc un séchage poussé de la résine pour ensuite la regonfler dans le solvant deutéré approprié avant d’entreprendre les expériences RMN. Les phases de séchage et de regonflement de la résine sont des étapes qui nécessitent beaucoup de temps et qui ne sont pas compatibles en vue d’une automatisation de ce type de technique. Nous avons donc utilisé le filtre de diffusion afin d’effectuer notre étude dans un solvant protoné. Les études effectuées précédemment au laboratoire ont montré que le filtre de diffusion est une séquence moins sensible qu’une expérience « single pulse » notamment par une perte de magnétisation pendant le temps d’application des gradients (l’aimantation se trouvant dans le plan xy), mais aussi par l’évolution de la constante de couplage J et enfin due aux imperfections des impulsions. Il est ŕ noter que ces facteurs ne sont pas constants pour toutes les molécules, comme par exemple la relaxation qui est plus longue pour le dernier résidu couplé[34]. Afin d’évaluer l’influence de la séquence LED sur notre systčme, nous avons tout d’abord utilisé cette séquence en solvant deutéré. Les résines contenant seulement un des deux composées nous ont permis de déterminer le facteur de correction ŕ appliquer pour utiliser le filtre de diffusion dans le cas des échantillons se trouvant dans le solvant protoné. Ce facteur de correction a été déterminé par calcul du rapport entre l’intégrale du Ha du pyroglutamate avec la moyenne des intégrales des protons alpha de l’alanine et de la phénylalanine dans le męme spectre de diffusion. Une fois ce rapport calculé, nous avons pu déterminer les différents pourcentages de la męme maničre que dans le cas précédent. Du fait de la moindre sensibilité du filtre de diffusion, nous n’avons pas pu déterminer les pourcentages des échantillons comptant respectivement 1% et 5% de pyroglutamate, dans les conditions que nous nous étions fixées (tableau1).
2.2.3 Limite de détection
Avec le développement de la chimie combinatoire, il est trčs important de disposer de méthodes de criblage de banques de molécules organiques. Les méthodes de criblage doivent ętre faciles ŕ mettre en śuvre, sensibles et rapides. De plus en plus les chimistes s’orientent vers une bille un composé (« one bead one compound »), il faut donc disposer d’une méthode d’analyse possédant une sensibilité suffisante pour pouvoir observer le composé ancré sur une bille de résine.
Le rayon des billes constituant les résines couramment utilisées en synthčse organique en phase solide est trčs variable pour un męme échantillon de résine et il est donc difficile d’estimer la charge d’une bille de ces résines. La société LCC quant ŕ elle fourni des résines dont le rayon des billes est hautement contrôlé et ce qui nous permet d’estimer la charge pour une bille de résine (figure 18).

Figure 18 : A gauche photo des billes d’une résine de Merrifield. A droite LCC dynosphčre
Si l’on considčre une résine de charge 0,4 mmol/g, on peut estimer que 1g de résine est constitué de 225 000 billes, ce qui conduit ŕ une charge par bille de l’ordre de 2 nanomoles. Cette charge trčs faible ne nous a pas permis de mettre en évidence les signaux de la molécule ancrée sur ce type de support. Pour surmonter ce problčme de charge insuffisante, nous avons utilisé des billes de résines dont la charge initiale ŕ été augmentée par un processus de dendrimérisation. La synthčse de ces billes consiste ŕ coupler sur les résines, des molécules polyfonctionnelles afin d’augmenter la charge initiale de la résine. En fonction du degré de substitution des différentes billes, on dispose de billes de charges différentes (schéma 2). Ces billes de charge élevée sont des équivalents des lanternes dont nous avons discuté au début de ce chapitre, puisque l’objectif est d’obtenir des objets aisément manipulables et possédant la charge la plus élevée possible. Les billes que nous avons utilisées ont été mises au point par le Dr Christophe Fromont au cours d’un stage post-doctoral dans le laboratoire du Pr Marc Bradley (Université de Southampton). La taille des billes utilisées était d’environ 500mm avec de grandes variations autour du rayon moyen. La charge des différentes billes variait de 50 nmol ŕ 180 nmol par bille.
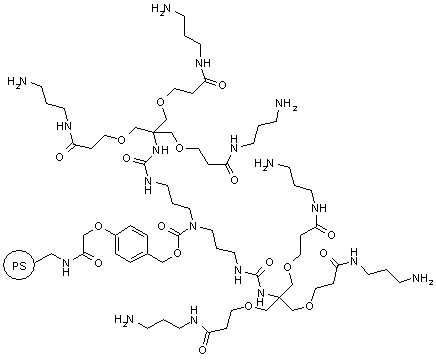
Schéma 2 : Formule développée plane d’un dendrimčre de 3éme génération de charge initiale 180nmol par bille.
Afin de développer une méthode de suivi de réaction nous nous sommes limités ŕ 64 scans pour l’enregistrement des spectres. Dans un premier temps nous avons couplé un acide aminé sur un lot de billes puis nous avons procédé ŕ une condensation de Wittig sur un dialdéhyde. Nous avons choisi d’effectuer une condensation de Wittig sur un dialdéhyde afin d’obtenir une résonance parfaitement isolée et clairement identifiable. L’enregistrement des spectres avec 64 scans (sur 5 billes chargées ŕ 180nmol) ŕ mis en évidence la présence de l’aldéhyde et l’enregistrement d’un spectre avec filtre diffusion nous a permis de savoir avec certitude que l’aldéhyde était parfaitement ancré sur le support solide (figure 17).
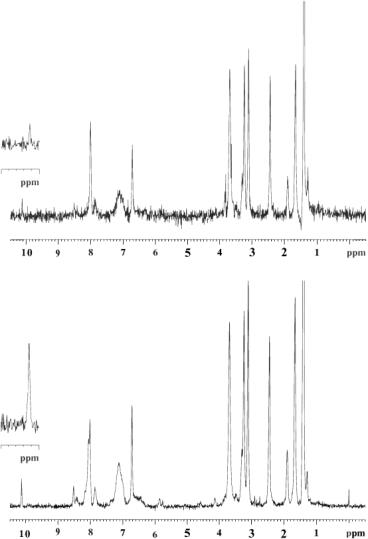
Figure 17 : Bas spectre enregistré sur 5 billes de dendrimčre (180nmol par bille) aprčs condensation de Wittig. Haut Spectre de diffusion du męme lot de bille.
Afin de pousser encore plus loin notre technique, nous avons tenté le suivi de réaction sur une bille individuelle pour connaître le seuil de détection en terme de charge de la RMN HRMAS. L’enregistrement en 64 scans des spectres sur les billes de charges 180 et 100 nmol, permet d’obtenir des spectres difficilement exploitables en utilisant une séquence d’impulsion classique, et l’utilisation du filtre diffusion ne nous permet pas d’obtenir des spectres de qualité suffisante pour procéder par la suite aux différents travaux d’interprétation. Les billes chargées ŕ 50 nmol ne permettent pas l’obtention de spectres exploitables que ce soit avec une séquence impulsionelle classique ou avec le filtre diffusion. Ce résultat est assez surprenant compte tenu des spectres obtenus par Duus et al 2000[35] sur des billes chargées ŕ 0,9 nmol. En effet, en 512 scans ils obtiennent des spectres parfaitement exploitables avec des largeurs de lignes proches de la RMN haute résolution liquide. En dehors du matériel utilisé pour l’acquisition des spectres qui ne peut expliquer ŕ lui seul une telle différence dans les résultats obtenus La différence de qualité des spectres obtenus peut s’expliquer par la structure des résines elles-męmes. En effet, de nombreuses études ont montré que la structure des résines et les solvants utilisés ont une grande influence sur la qualité des spectres. Une étude récente[36] a montré que les résines entičrement basées sur du polyéthyléneglycol présentent un meilleur gonflement que les résines basées sur du divinylbenzčne. Le gonflement des résines étant étroitement lié ŕ la mobilité des molécules ancrées sur le support solide, cela se traduit immédiatement sur la qualité du spectre en terme de largeur de lignes, mais le paramčtre qui contribue le plus a l’élargissement des lignes est sűrement la présence d’aromaticité dans la structure de la résine.[37] La présence des cycles aromatiques dans la structure de la résine est ŕ l’origine d’un champ magnétique local induit par le mouvement des électrons. Un proton situé au voisinage des ces cycles aromatiques va donc ressentir un champ magnétique différent suivant l’orientation des cycles. La variation du champ magnétique local autour du proton va donc induire des déplacements chimiques différents qui conduisent ŕ l’élargissement des lignes. Dans leur étude Duus et al ont utilisé des billes de résine POEPOP1500. Ce type de résine, comme nous l’avons déjŕ vu précédemment, est particuličrement favorable pour une étude RMN HRMAS, puisque ces résines sont totalement dépourvues de cycles aromatiques. Le relatif échec que nous avons rencontré pour déterminer la limite de détection en terme de charge de la RMN HRMAS, est d’une part lié au type de support utilisé, mais aussi aux conditions d’acquisition. En effet, nous nous sommes limités ŕ 64 scans pour développer une méthode permettant le criblage ŕ haut débit, alors que Duus et al, ont utilisé 8 fois plus de scans. Toutefois la différence de qualité entre les spectres que nous obtenus sur les dendrimčres et la qualité des spectres obtenus sur des billes de POEPOP1500 n’est pas uniquement liée au nombre de scans mais plus particuličrement au support utilisé.
3 Conclusion
Dans ce chapitre nous avons vu que la RMN HRMAS peut apporter une réponse intéressante aux problčmes rencontrés pendant la SOPS. Cette technique permet d’une part d’étudier les nouveaux supports développés pour la synthčse organique en phase solide, et permet aussi de suivre des transformations rapides sur ce męme de type de support, sans nécessité de séparer les molécules du support, mais aussi sans aucune modification de l’échantillon. De plus, nous avons aussi montré que la RMN HRMAS était une technique sensible pour déterminer précisément le pourcentage d’impuretés formées au cours d’une réaction chimique sur différents supports solides. Toutefois cette technique, malgré sa grande efficacité pour étudier des milieux hétérogčnes, est limitée par sa sensibilité en terme de charge et la qualité des spectres obtenus est hautement liée au support utilisé.