Chapitre I
Comparaison synthèse en phase solide et synthèse en phase
homogène
1 Principes des deux modes de synthèse
1.1 Principe général de la synthèse en phase homogène
La synthèse en phase homogène est utilisée depuis la nuit des temps. En effet les premières traces de réactions chimiques remontent à 400 000 ans (combustion). Comme son nom l’indique, la synthèse en phase homogène consiste à mettre en présence deux espèces réactives dans la même phase. En solution, les molécules sont libres de se déplacer par diffusion moléculaire et des collisions peuvent se produire. Dans le cas où la géométrie et les charges électrostatiques respectent les fondements de base de la chimie (ex : addition d’un nucléophile sur un électrophile), une nouvelle liaison peut se former et une nouvelle molécule est obtenue. Bien que de mise en œuvre facile, la limitation de cette technique est qu’elle nécessite une purification entre chaque étape lors d’une synthèse multi-étape.
1.2 Principe général de la synthèse en phase solide
Depuis son introduction en 1963 par Merrifield[1] dans le cadre de la synthèse peptidique, la synthèse en phase solide a connu un essor considérable, notamment avec l’introduction de la chimie combinatoire. Cette technique repose sur un principe simple mais ingénieux qui consiste à immobiliser un des réactifs sur un polymère insoluble. Ce procédé permet alors d’utiliser de larges excès de l’autre réactif sans problème d’élimination en fin de réaction. La simplicité de mise en œuvre de la technique a permis une automatisation rapide en particulier pour des chimies à caractère répétitif, comme la synthèse des peptides ou d’oligonucléotides. Malgré sa facile mise en œuvre et une automatisation aisée dans ces deux applications, la synthèse en phase solide appliquée à la chimie organique en général, connaît de nombreuses limitations. Cet aspect sera discuté plus loin.
2 La synthèse peptidique
2.1 Synthèse peptidique en phase homogène
La synthèse peptidique consiste en une suite de réactions (formation d’un lien amide) entre différents acides aminés pour obtenir une molécule plus complexe : un peptide. Avant l’introduction de la phase solide, les synthèses peptidiques étaient réalisées en phase homogène. Deux méthodologies étaient alors utilisées. La première méthode consistait en la condensation de fragments. Un octapeptide pouvait être obtenu par condensation de deux tétrapeptides eux-mêmes obtenus à partir de deux dipeptides. La méthode de synthèse par couplage de fragments est particulièrement utilisée pour la synthèse de peptides longs ou encore pour la synthèse de séquences dites difficiles. L’avantage majeur de cette méthode est que d’une part les différents fragments intermédiaires sont purifiés et caractérisés, mais surtout, les propriétés physico-chimiques des différents fragments sont très nettement différentes du peptide désiré ce qui facilite les étapes de purification. Une autre approche consistait à synthétiser le peptide pas à pas. Une des premières synthèses de peptide par cette approche remonte à 1953. A cette époque Du Vigneau et collaborateurs[2] avaient réussi la synthèse de l’ocytocine, une hormone peptidique de neuf acides aminés. A première vue, la synthèse peptidique par cette stratégie peut paraître simple, mais il ne faut pas négliger que les différents acides aminés comportent des fonctionnalités différentes qui peuvent conduire à de nombreux produits secondaires. Prenons le cas du couplage d’une glycine sur l’alanine. Lors de la mise en réaction de ces deux composés, on peut obtenir un mélange de produits composés de H-Ala-Ala-OH, H-Gly-Gly-OH, H-Ala-Gly-OH (peptide désiré), mais aussi H-Gly-Ala-OH. Cette stratégie de synthèse implique donc des étapes de purification et d’analyse entre chaque couplage et la complexité du mélange augmente lorsque les acides aminés utilisés possèdent des chaînes latérales réactives. Une des possibilités pour éviter ce genre de mélange complexe est le recours à des esters activés[3],[4]. L’utilisation des esters activés permet de diminuer la complexité du mélange en favorisant la formation du produit désiré. On peut également diminuer la complexité du mélange en recourant à une protection de la fonction aminea ce qui permet d’obtenir uniquement le produit désiré. Lorsque les acides aminés utilisés possèdent une chaîne latérale réactive (ex : lysine), il faut également protéger la chaîne latérale afin d’éviter son implication dans la réaction. Ce type de synthèse ne permet pas l’utilisation d’un large excès de réactif, puisque tout excès de réactif devient une impureté pour le produit final. La stratégie de synthèse peptidique pas à pas en phase homogène, bien qu’efficace, reste très contraignante, car elle nécessite une étape de purification après chaque étape.
2.2 Synthèse peptidique en phase solide
L’introduction de la synthèse en phase solide a révolutionné la synthèse peptidique. En 1963 Merrifield a eu l’idée géniale d’ancrer le premier acide aminé à une matrice de polymère insoluble. Le protocole de synthèse pas à pas, développé précédemment, pouvait ensuite être appliqué (figure 1). Un premier avantage de la synthèse peptidique en phase solide est que le composé intermédiaire d’intérêt, est ancré sur la phase solide, ce qui supprime les étapes de purification car un simple lavage suffit pour éliminer les réactifs restés en solution. Le second avantage de l’ancrage de molécules sur un support solide, qui découle du premier est que la synthèse sur support solide permet l’utilisation d’un excès de réactifs, puisque la molécule d’intérêt reste sur le support solide jusqu'à la fin de la synthèse et à nouveau l’excès peut être éliminé par un simple lavage. La synthèse de peptides consiste donc en une répétition d’étapes de couplages et de déprotections jusqu'à obtention du peptide désiré, qui est ensuite séparé du support par coupure chimique et ensuite purifié et analysé.

Figure 1 : Représentation simplifiée de la synthèse peptidique en phase solide.
C’est la répétitivité et l’efficacité de la chimie des peptides qui a permis l’adaptation rapide sur la phase solide. Non seulement l’adaptation de la synthèse peptidique en phase solide a été aisée, mais en plus, la simplicité de la chimie (répétition de cycles) a contribué à son automatisation rapide. Lors de la synthèse peptidique, deux groupements protecteurs sont à notre disposition pour protéger la fonction amine a. Ces deux types de protection temporaire sont dits orthogonaux. La principale différence entre les deux groupes protecteurs vient du mode de déprotection. Le groupement Fmoc actuellement le plus utilisé est déprotégé par utilisation de pipéridine diluée (20% v/v DMF, milieu basique). Le groupement Boc est quant à lui déprotégé par utilisation de TFA (milieu acide), son utilisation nécessitant donc une étape de neutralisation avant de pouvoir réaliser le couplage suivant. Pour chaque type de protection de la fonction amine a, sont associés des groupements protecteurs pour la protection des chaînes latérales. Ces groupements protecteurs dits « définitifs » car ils sont résistants dans les conditions de déprotection de la chaîne principale, ne seront enlevés que lors de la séparation du peptide du support solide.
Malgré sa grande efficacité, la synthèse de peptides en phase solide pose des problèmes pour certaines séquences peptidiques. Considérons tout d’abord le problème du rendement pour la synthèse de longues séquences. Lors de synthèses en phase solide, les rendements de couplages sont de l’ordre de 99% dans le meilleur des cas. Pour un peptide de 50 acides aminés, le rendement final de synthèse est alors fortement réduit et si le rendement de couplage par acide aminé est de 90%, le rendement final de synthèse est catastrophique. De plus, chaque délétion conduit à un grand nombre de peptides de longueurs différentes, ce qui conduit à un mélange complexe, difficile à purifier. La complexité d’un tel mélange peut être réduite par acylation de l’amine libre (blocage irréversible : « capping ») après chaque étape de couplage, ce qui permet d’éliminer les peptides dans lesquels il manque un ou plusieurs acides aminés à l’intérieur de la séquence.
En dehors des séquences longues, il existe certaines séquences pour lesquelles les problèmes débutent pour des longueurs inférieures à 10 acides aminés en particulier entre 5 et 10 AA. Ces séquences ont été appelées « séquences difficiles » et sont caractérisées par des rendements très faibles en peptides après séparation du support. [5],[6],[7] Ces faibles rendements de synthèse sont dus à des couplages et surtout à des déprotections incomplètes, mais quel est le phénomène à l’origine de ces difficultés ? De nombreuses hypothèses ont été émises pour essayer de comprendre les difficultés de synthèses rencontrées. Une première hypothèse consistait à dire que la résine avait atteint sa limite de charge due à une gêne stérique importante, mais cette hypothèse a été rapidement réfutée par Merrifield et collaborateurs.[8] Lors de cette étude Sarin et al ont synthétisé un peptide test (LAGV) sur une résine aminométhyl en faisant varier la longueur du « spacer » et en faisant également varier le degré de substitution de la résine. Les résultats obtenus lors de cette étude montrent que ni la longueur du « spacer » ni la charge de la résine n’ont une influence significative sur le rendement et la pureté du peptide obtenu. Une autre interprétation de ces chutes de rendements de synthèses pour des peptides longs a été proposée par Tam et al.[9] En effet, en utilisant une résine de Merrifield (résine polystyrène), ils ont mis en évidence une transition de polystyrène vers polyamide dans la nature physico-chimique de la peptidyl résine qui évolue d’un caractère hydrophobe vers un caractère hydrophile lorsque la charge en peptide augmente. Lors de cette étude, un peptide de 8 acides aminés a été synthétisé sur une résine de Merrifield dont la charge avait été augmentée à l’aide d’arbres de Lysine. Cette transition de polystyrène vers polyamide due à l’augmentation de la quantité de peptide conduit à un changement important dans la nature du système impliquant des problèmes de solvatations. Si l’on ne change pas de solvant la disponibilité des réactifs diminue, ce qui conduit inévitablement à des chutes de rendements. Lorsque la charge en peptide augmente pour atteindre un rapport de 50% par rapport à la résine, celle-ci adopte le comportement d’une résine polyamide.
Une autre hypothèse mettait en cause la formation d’une structure secondaire intra-chaîne. Comme il est clair que ce phénomène peut avoir une influence directe sur la synthèse du peptide, il est très important de déterminer quels sont les facteurs qui peuvent influencer la structuration d’un peptide lorsque celui-ci est encore ancré sur le support solide, tel que la nature des groupes protecteurs utilisés, l’influence de la charge de la résine ou encore la structure du polymère. Néanmoins le problème lié à la structuration est difficile à vérifier avec la méthode classique qui consiste à séparer le peptide du support solide et par la suite, procéder à son analyse par les méthodes classiques comme l’HPLC, la RMN, ou encore la spectroscopie de masse. Le problème de cette méthode d’analyse est que seul le peptide est étudié alors que les problèmes sont liés à la paire peptide protégé/résine. Au cours d’études précédentes, il a été montré que l’utilisation de la stratégie Boc conduisait à une moins grande chute de rendement que l’utilisation de la stratégie Fmoc.5 Cette constatation a été expliquée par la formation de liaisons hydrogènes entre le peptide et les groupements aromatiques de la résine lorsque la fonction aminea était sous forme NH2. La formation de ces liaisons hydrogènes a été mise en évidence lors d’une étude par spectrométrie IR.5 Ces liaisons hydrogènes contribuent d’une manière non covalente à une réticulation plus importante des résines ce qui conduit à un moindre gonflement.[10] Une réticulation supplémentaire des résines empêche une bonne diffusion des réactifs dans la matrice solide, ce qui a une influence directe sur les cinétiques de couplages et de déprotections. Le même type de corrélation peut être effectué par RMN HRMAS, où une corrélation directe entre la largeur des signaux obtenus et l’état de protonation N-terminal du peptide a été observée.[11] Lorsque le N-terminal du peptide est sous forme NH3+ (cas de la chimie Boc), la résine gonfle de façon importante et les signaux obtenus sont fins, par contre dans le cas de la chimie Fmoc, le N-terminal est sous forme NH2 après déprotection et les signaux obtenus sont fortement élargis, ce qui va dans le sens d’une réticulation plus importante des résines.
Une meilleure compréhension des phénomènes internes qui conduisent à l’agrégation des peptides est donc primordiale pour l’avancement de la synthèse peptidique en phase solide. Il existe des techniques colorimétriques pour déterminer l’efficacité des couplages ou des déprotections, mais aucune technique ne permet le suivi pas à pas au niveau de la structuration du peptide lors de la synthèse. L’amélioration des conditions de la synthèse peptidique pour les séquences dites difficiles passe inévitablement par une étude de la structuration de peptides modèles sur différents supports solides. Peu de méthodes ont été développées pour suivre la croissance de la chaîne peptidique en formation. En incorporant des noyaux de deutérium au niveau de l’acide aminé (sur le groupement protecteur), l’IRTF[12] permet d’évaluer le taux de déprotection lors de la synthèse de peptides. L’incorporation d’un acide aminé non naturel contenant un radical libre, combiné à la RPE,[13] permet d’évaluer la dynamique macroscopique des chaînes. Mais, pour des peptides en absence de marquage, il existe peu de méthodes pouvant fournir une vision structurale détaillée pendant toute la synthèse. Notre groupe avait déjà appliqué la RMN HRMAS à l’étude de la synthèse peptidique d’une séquence difficile et avait montré dans la cas de l’Aggrécan que la difficulté de synthèse de ce peptide de 10 acides aminés était due à l’agrégation du peptide.11 La corrélation entre la chute de rendement et l’élargissement des raies suggérait que la perte de mobilité des chaînes était due soit à leur interaction mutuelle soit à leur interaction avec la résine. Une autre étude concernant la synthèse du poly-alanine, séquence difficile par excellence, a montré que cet élargissement provenait bien de l’agrégation des chaînes.[14] La dilution des chaînes sur la résine, qui par sa nature rigide permet de séparer physiquement les chaînes peptidiques et donc d’empêcher l’agrégation, avait permis d’obtenir les peptides avec d’excellents rendements et d’observer la transition structurale des chaînes d’une conformation aléatoire vers une structure en hélice a à partir d’une longueur de 12 résidus. Cette dernière observation suggère que les problèmes de synthèse sont dus à l’absence de structure secondaire stable permettant une agrégation du peptide pendant la synthèse. Ce même phénomène est observé avec des peptides ou des protéines mal repliés dans la cellule, qui en s’agrégeant peuvent être à la base d’un disfonctionnement cellulaire. Nous avons voulu observer de tels peptides sur un support solide, mais afin de maintenir le contexte biologique, nous avons utilisé des résines compatibles avec un gonflement dans un milieu aqueux. Récemment il a été démontré que des peptides pouvaient adopter une structure en hélice alpha sur des résines compatibles avec un milieu aqueux[15]. Au laboratoire nous avons appliqué la RMN HRMAS à l’étude de la structuration ou non d’un peptide modèle de feuillet b intramoléculaire sur différents supports solides hydro-gonflables.
3 Synthèse d’oligonucléotides
3.1 Synthèse d’oligonucléotides en phase homogène
Depuis la découverte de la fonction de l’ADN ainsi que la détermination de sa structure, de nombreux efforts ont été développés pour obtenir des brins d’ADN de façon synthétique. L’ADN consiste en une chaîne de 2-déoxy-D-riboses reliés entre eux en 3’-5’ par un lien phosphodiester. Chaque unité de ribose porte en position 1’ une des quatre bases possibles (A, T, G, C). La première synthèse d’un oligonucléotide de façon synthétique a été réalisée en 1955 par Michelson et al[16], utilisant un phosphotriester. Cette méthode a été très longtemps utilisée pour la synthèse des oligonucléotides en phase homogène. Toutefois la synthèse d’oligonucléotides en phase homogène reste complexe. La présence de nombreuses fonctions réactives (fonction OH sur le sucre, amine primaire sur certaines bases), nécessite l’utilisation de groupements protecteurs pour éviter les réactions secondaires.
3.2 Synthèse d’oligonucléotides en phase solide
Juste après l’apparition de la synthèse en phase solide pour la chimie des peptides, le concept a rapidement été adapté par Letzinger[17] pour la synthèse d’oligonucléotides. La synthèse d’oligonucléotides sur la phase solide repose sur les mêmes concepts que pour les peptides. L’oligonucléotide en croissance, reste ancré sur le support jusqu’à la fin de la synthèse, ce qui permet de nouveau l’utilisation d’un large excès de réactif pour effectuer une conversion totale. La répétitivité des cycles permet une automatisation de la procédure. Le support solide utilisé pour la synthèse d’oligonucléotides est toutefois légèrement différent de celui utilisé en synthèse peptidique. Le support principalement utilisé est le support CPG (Controlled Pore Glass). Ce support consiste en une matrice de verre à pores hautement contrôlés, généralement dérivée par une longue chaîne alkyl amine. Ici encore, nous retrouvons une protection temporaire et une protection permanente. Dans le cas de la synthèse des oligonucléotides, le groupement temporaire se trouve en 5’ sur le sucre, il s’agit d’un groupement trityl et les amines des bases sont protégées par un groupement benzoyle pour l’adénine et la cytosine, alors que la thymine est protégée par un groupement isobutyryl. La synthèse d’oligonucléotides consiste en une suite de cycles de déprotection et couplage. Le produit désiré est ensuite séparé du support en utilisant de l’ammoniaque (Figure 2).
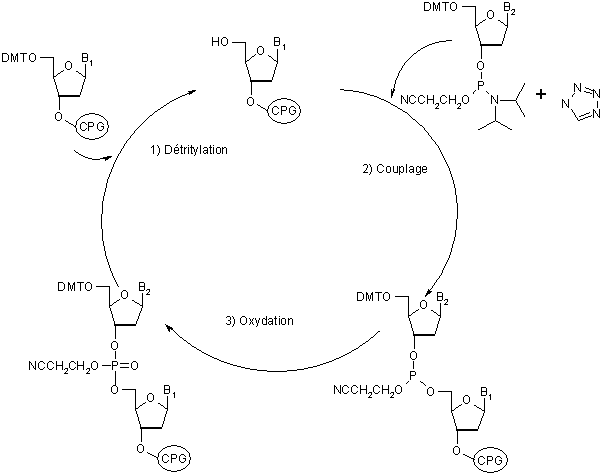
Figure 2 : Représentation cyclique de la synthèse en phase solide d’oligonucléotides par la méthode phosphitetriester.
4 Synthèse en phase homogène et synthèse en phase solide en chimie organique
Alors que la synthèse peptidique a pu s’adapter très facilement à la phase solide de part la simplicité de la chimie utilisée et la répétitivité de cycles, l’adaptation de la synthèse organique est beaucoup plus difficile. Cette difficulté provient d’une part de la diversité des réactions à adapter, du manque de techniques d’analyses, mais aussi de par la nature du polymère utilisé.
Au cours de cette partie, les différents aspects de la synthèse en phase homogène et de la synthèse en phase solide seront mis en opposition vis à vis de leurs apports respectifs à la chimie organique, domaine pour lequel les différences entre les deux modes de synthèses sont les plus marquées. Les points abordés au cours de cette partie seront les points considérés comme les plus importants de l’article de revue de Baldino[18]. Dans cette revue Baldino discute les principales différences entre la synthèse en phase solide et la synthèse en phase homogène. Les point abordés dans cet article nous servirons de base pour la suite de la discussion.
4.1 Nombre de réactions accessibles
Le point qui est à mon sens une des oppositions majeures des deux modes de synthèse est le nombre de réactions accessibles. La synthèse en phase homogène étant utilisée depuis plus longtemps que la Synthèse Organique en Phase Solide (SOPS), le nombre de réactions décrites à ce mode de synthèse est beaucoup plus important. Leur transfert vers la phase solide demande souvent une étape supplémentaire de mise au point qui limite encore actuellement son application plus large. Ceci était en particulier dû à la présence du support solide lui-même et aussi à cause du manque de techniques d’analyses permettant de suivre une transformation chimique directement sur le support. Malgré ces deux facteurs limitants, depuis ces dix dernières années, le nombre de réactions adaptables à la chimie en phase solide est en constante augmentation.[19],[20],[21] L’augmentation du nombre de réactions adaptées à la phase solide est liée à l’efficacité synthétique de la technique qui permet de conduire les réactions jusqu’a leur terme. Cette caractéristique est particulièrement intéressante lors de la synthèse de molécules complexes qui implique un grand nombre d’étapes. Dans le cas d’une synthèse courte en phase homogène (par exemple 3 étapes), le temps investi est principalement consacré au temps de réactions et pas dans les étapes de purification. Par contre dans le cas de la synthèse d’une molécule complexe (en phase homogène), le temps investi dans les étapes de purification est considérable. La synthèse en phase solide est dans ce cas très utile puisqu’une seule étape de purification est réalisée en fin de synthèse, les purifications intermédiaires étant remplacées par un simple lavage. Mais le principal frein au développement de la synthèse en phase solide est le manque d’informations sur la nature du support utilisé et surtout la grande variabilité des caractéristiques physico-chimiques des différents lots d’une même résine
4.2 Choix des groupes protecteurs
Dans le cas de la synthèse en phase homogène, le choix des groupes protecteurs est limité aux groupements chimiques dont le produit de déprotection conduit à des résidus volatiles tel que le groupement Boc qui conduit à du CO2 et de l’isobutène. Depuis quelques années, le choix des groupes protecteurs pour la synthèse en phase homogène s’est agrandi avec l’introduction des résines « scavenger » qui permettent de piéger les résidus de déprotection, en permettant ainsi une séparation facile du composé désiré des différentes impuretés. Dans le cas de la synthèse en phase solide, le choix des groupes protecteurs est beaucoup plus important grâce à la caractéristique principale de la phase solide qui permet des lavages abondants après chaque étape et ne se limite donc pas l’utilisation de groupements protecteurs dont les résidus de déprotection sont volatils.
4.3 Choix des solvants et de la température.
Lors d’une synthèse en phase homogène, on dispose d’un large choix de solvants pour effectuer une transformation chimique. Il n’existe en effet aucune limitation quant au choix du solvant, hormis un problème de solubilité des réactifs. Dans le but d’une synthèse automatisée, le choix du solvant peut toutefois connaître quelques limitations, les systèmes automatisés n’étant pas compatibles avec les solvants très volatiles comme l’éther éthylique ou encore le dichlorométhane. Une autre limitation que peut connaître l’automatisation concerne la distribution de petits volumes.
En ce qui concerne la phase solide, le problème du solvant est tout autre. De part la nature du polymère, tous les solvants ne sont pas utilisables. Certains solvants sont incompatibles avec la nature physico-chimique des résines et peuvent déformer voir détruire le support solide. Les solvants les plus « classiquement » utilisés en phase solide pour obtenir un gonflement approprié d’un support basé sur du polystyrène réticulé sont : DMF, NMP, THF, DCM, MeOH. D’autres solvants, peuvent conduire à un rétrécissement de cette même matrice solide et ainsi empêcher la bonne pénétration des réactifs dans la matrice solide. L’eau et le DMSO ne gonflent pas les résines de type polystyrène et ces deux solvants ne sont utilisables que depuis l’apparition des résines à base de polyéthylène glycol ou de polyacrylamide.
Une autre grande différence entre les deux modes de synthèse est la gamme de températures utilisables. En disposant du matériel approprié, la synthèse en phase homogène ne connaît aucune limitation pour le choix de la température. De façon générale, on peut dire que la gamme de températures accessibles à la synthèse en phase homogène peut varier de +200°C à –80°C. La synthèse en phase solide connaît une contrainte supplémentaire liée au caractéristiques physico-chimiques du support solide. Dans le cas d’une résine polystyrène réticulée à 1% avec du divinylbenzène, la température de transition vitreuse se situe autour de 100°C. La température de transition vitreuse est la température à laquelle les caractéristiques physico-chimiques de la résine changent. Compte-tenu de ce phénomène, il est donc impossible d’effectuer une réaction à une température supérieure à la température de transition vitreuse, la zone dite de confort se situant même en dessous de 80°C. A l’opposé, des réactions nécessitant de basses températures comme la génération d’énolates sur phase solide, se réalisent très facilement.[22], [23]
4.4 Coût des réactifs et du matériel
La synthèse en phase homogène encourage la parcimonie pour la quantité de réactifs à utiliser, car tout excès de réactif peut devenir une impureté pour le produit final et ainsi modifier les résultats lors du criblage de la banque. La SOPS, quant à elle, encourage l’utilisation d’un excès de réactif pour conduire la transformation chimique jusqu’à son terme, et nécessite également une grande quantité de solvant (lors des étapes de lavage) ce qui contribue à augmenter le coût de la synthèse. De plus, si l’on ajoute le coût des résines[24] et l’investissement pour obtenir le matériel spécialisé comme les réacteurs à usage unique, le développement d’une synthèse en phase solide peut devenir très onéreux. Pour la synthèse d’un peptide de 34 acides aminés (échelle 0.1 mmole) sur un synthétiseur en flot continu en simple couplage, il faut compter 101€ pour les acides aminés, 108€ pour les solvants (DMF) et 25€ pour la résine (NOVASYN TG). Dans le cas de certaines bibliothèques de molécules, il faut encore rajouter la nécessité de développer un «linker» complexe, ce qui peut encore contribuer à augmenter le coût de la synthèse.
4.5 Re-synthèse de molécules biologiquement actives à large échelle.
Dans le cas de la synthèse en phase homogène, la re-synthèse de molécules biologiquement actives n’est pas un problème trop difficile à surmonter, mis à part les problèmes de transition d’échelle (scale up).
En ce qui concerne la phase solide, le problème est tout autre. Un des premiers problèmes rencontrés est la nécessité d’obtenir une grande quantité d’un même lot de résine pour éviter les problèmes de reproductibilité d’un lot de résine à un autre. En général, la synthèse à large échelle de composés biologiquement actifs mise au point en phase solide est produite en phase homogène. La synthèse en phase solide est souvent limitée à la mise au point initiale d’une synthèse et reste limitée à des synthèses à l’échelle de quelques milli-moles. Cette limitation d’échelle est notamment liée au coût important des résines mais aussi au coût des solvants et des réactifs car la synthèse en phase solide, pour aller à son terme, nécessite l’utilisation de gros excès. En dehors des coûts des différents matériels, les synthèses sur des quantités importantes de résines exigent une grande quantité de solvant pour permettre une bonne diffusion des réactifs dans toute la résine et de tels mélanges sont difficiles à agiter correctement.
4.6 Méthodes de contrôle pendant la synthèse
Afin d’optimiser une réaction chimique sur le support solide, il est indispensable de disposer d’outils qui permettent de contrôler l’avancement des réactions. Dans le cas de la synthèse en phase homogène, un grand nombre de techniques est disponible pour suivre l’avancement d’une réaction, l’expérimentateur pouvant utiliser la CCM, L’HPLC, la RMN, la spectroscopie de masse ou encore l’IR. Par contre, l’analyse d’une synthèse en phase solide est limitée. De manière générale, l’expérimentateur ne dispose que de tests colorimétriques qui ne sont pas quantitatifs. La seule possibilité pour suivre de manière quantitative une réaction chimique sur le support solide consiste à séparer la molécule de son support par une coupure chimique irréversible, pour ensuite procéder à l’analyse. Cette méthode, qui permet une analyse quantitative et un suivi pas à pas, est néanmoins lourde à mettre en œuvre et a longtemps freiné la mise au point de réactions sur support solide. Elle est très contraignante car elle nécessite beaucoup trop de temps dans le but d’optimiser une réaction, de plus la coupure chimique peut dans certains cas entraîner la formation de produits secondaires. La transformation du Glutamate en Pyroglutamate en milieu fortement acide sera étudiée au cours du deuxième chapitre pour explorer la limite de détection d’impuretés de la RMN HRMAS au cours d’une synthèse en phase solide. Il est donc primordial de disposer d’outils permettant de suivre de façon rapide et précise une réaction chimique sur un support solide, sans nécessiter de coupure. Depuis quelques années de nombreux efforts ont été développés pour mettre au point des méthodes permettant l’analyse directe d’une molécule ancrée sur le support. Ces méthodes sont entre autres la FTIR, la RMN gel 13C, ou la spectroscopie de masse. La RMN HRMAS qui est au cœur de nos travaux, a déjà montré sa capacité pour le suivi de réactions comme lors de la formation d’une liaison carbone-carbone sur le support solide[25]. Lorsque l’on veut suivre une réaction sur un support solide, il convient de s’interroger sur le but de l’analyse. Suit-on la réaction dans le but de mettre en évidence la formation de produits secondaires, veut-on identifier la molécule formée, ou encore déterminer le pourcentage de produit formé ? En fonction de la question posée l’utilisateur dispose de différentes techniques d’analyses, chacune apportant sa spécificité. Luo et al ont montré dans le cas du suivi d’une réaction en cinq étapes que la FTIR et la RMN HRMAS sont deux techniques complémentaires.[26]
4.7 Rôle du support solide
Après les considérations techniques précédentes concernant l’utilisation de la phase solide pour la chimie, il convient de se poser quelques questions sur le support solide lui-même. Pendant des années, les chimistes ont souvent considéré le support solide comme « inerte » vis à vis des transformations chimiques. Longtemps les articles traitant de chimie en phase solide considéraient le support solide comme de petites sphères avec des molécules attachées dessus. C’est à la fin des années 90 que les chimistes ont commencé à se poser des questions sur l’influence de la résine sur l’évolution de la réaction[27].
Un des points qui est à mon sens le plus important à aborder est la relation solvant-résine. Dans l’esprit de beaucoup, le paramètre le plus important dans la mise au point d’une réaction sur support solide, est le choix du solvant alors que dans la réalité il faut prendre en compte la paire solvant-support. Il est vrai que si le solvant choisi ne conduit pas à un gonflement suffisant de la résine, la pénétration des réactifs ne sera pas efficace, mais il ne faut pas perdre de vue que la transformation chimique se passe dans l’ensemble solvant-résine : c’est le support gonflé qui est le vrai solvant de la réaction. De la même manière qu’une meilleure connaissance des solvants (ex : protique, aprotique, polaire, apolaire) utilisés en chimie en phase homogène a permis une amélioration des réactions, une meilleure connaissance de la résine gonflée permettra certainement une amélioration de la synthèse en phase solide. Une des meilleures illustrations de l’importance de la dualité solvant-résine, est qu’au cours d’une synthèse, les molécules ancrées sur le support sont solubles dans le solvant utilisé alors que la même molécule prise individuellement n’est pas soluble dans ce même solvant[28]. Lors d’une expérience illustrant ce phénomène, un peptide dérivé de la protéine M603 a été synthétisé sur la phase solide à l’aide de dichlorométhane comme solvant mais lors de la coupure du peptide protégé par irradiation celui-ci était insoluble dans la majorité des solvants. Une autre illustration de cette dualité solvant-résine peut être constatée en suivant la cinétique d’une même réaction sur deux supports ne possédant pas le même gonflement dans un solvant donné. Dans bien des cas, la cinétique de réaction n’est pas directement reliée au taux de gonflement de la résine. Ce résultat peut paraître surprenant sachant qu’il est couramment acquis que 99% des points de fixation des molécules se trouvent à l’intérieur des billes, bien que ce résultat soit toujours soumis à controverse. De nombreuses méthodes ont été développées pour quantifier le gonflement du support dans divers solvants[29],[30],[31]et ont ainsi conduit à l’actuelle gamme de solvants utilisables en synthèse organique en phase solide.
Un autre paramètre à prendre en compte lors d’une synthèse en phase solide est que la présence de la molécule greffée sur le polymère, change les propriétés thermodynamiques du polymère, comme les propriétés de gonflement par exemple. En 1980 Sarin et al[32] ont couplé un pourcentage variable de peptides sur une résine et ont observé la variation de volume d’une bille de résine par rapport à une bille de résine sèche dans différents solvants. Les résultats obtenus montrent bien l’influence de la présence du peptide ancré sur les propriétés thermodynamiques du polymère. Une bille de résine nue gonflée dans le dichlorométhane présentait une taille 1,84 fois plus importante que la résine sèche, alors que la même bille de résine chargée en peptide présentait cette fois une taille seulement 1,33 fois plus importante. A l’opposé, la même étude menée dans le DMF conduit à des résultats inverses. Cette expérience montre qu’il n’est pas toujours judicieux d’effectuer une suite de réactions dans le même solvant, même si le solvant utilisé dans la première étape était le solvant le plus approprié.
Une autre caractéristique importante du support solide concerne le degré de réticulation ainsi que le mode de synthèse des différentes résines. La variabilité du degré de réticulation du support solide a souvent contribué à freiner son utilisation pour la production de molécules à large échelle. Il existe actuellement deux modes de synthèse de support solide : la polymérisation par suspension et la polymérisation « en vrac » (bulk polymérisation). La polymérisation en vrac consiste à chauffer les différentes monomères en présence d’un initiateur. Le polymère ainsi obtenu est ensuite moulu et tamisé afin d’obtenir des particules de la même taille. La polymérisation par suspension est réalisée dans un système biphasique constitué par les monomères (phase1) suspendu dans un (non)solvant (phase2). Un bon contrôle de l’agitation de la suspension permet d’obtenir des particules de diamètre contrôlé. La polymérisation par suspension est la méthode la plus utilisée pour l’obtention de supports solides. Ce mode de synthèse des supports solides ne permet pas toujours un bon contrôle du degré de réticulation des polymères et conduit souvent à des inhomogénéités dans la résine qui peuvent donner lieu à une non reproductibilité de certaines réactions sur les différents supports solides[33]. Prenons le cas d’une résine Merrifield obtenue par polymérisation du styrène et de divinylbenzène, les groupements chloro-méthyle étant introduits par co-polymérisation avec du chlorovinylbenzène. Le divinylbenzène se trouve à l’état de mélange de deux isomères méta et para, ces différents isomères n’ayant pas la même réactivité lors de synthèse de la résine de Merrifield. Des études sur les polymères ont montré que l’isomère méta du divinylbenzène réagit plus vite que l’isomère para[34]. Si l’on ajoute à cela le changement de réactivité du divinylbenzène après la formation du premier point de réticulation, on constate les difficultés importantes qui sont rencontrées pour obtenir une résine parfaitement homogène.
Une autre source d’inhomogénéité lors de la synthèse de résine provient des solvants utilisés pour éliminer les monomères n’ayant pas réagi. Suivant les solvants utilisés pour éliminer les monomères en excès, la taille des pores obtenus est sensiblement différente. Les résines séchées à partir de « mauvais » solvants conduisent à une porosité maximum alors que les résines séchées à partir de bons solvants conduisent à une moindre porosité[35].
Compte-tenu des différents problèmes soulevés par le support solide (gonflement, interaction solvant-résine, réticulation), nombres de techniques spectroscopiques ont été développées pour apporter certaines réponses. La RMN et la RPE permettent d’apporter des réponses concernant la dynamique du support, alors que les techniques basées sur la fluorescence ou encore l’Infra Rouge se consacrent à l’étude d’un état statique de la résine.
Un nombre important de techniques a été développé pour étudier les différentes caractéristiques des supports solides utilisés en chimie combinatoire, mais aucune technique prise individuellement ne permettra de comprendre un système aussi complexe qu’une matrice solide, chaque technique apportant sa contribution pour une meilleure compréhension de tels systèmes. Malgré le grand nombre de réponses apportées par ces différentes techniques, il subsiste de nombreuses questions notamment en ce qui concerne le contrôle de la réticulation des différents polymères.
5 La Chimie Combinatoire : définitions et utilisations
5.1 Principe de la Chimie Combinatoire
Traditionnellement la mise au point de molécules à visée thérapeutique se fait par optimisation de la structure d’une molécule de référence essentiellement dérivée des produits naturels ou par modification de la structure d’une molécule prototype, déterminée lors du criblage initial.
Plus récemment, le développement de nouvelles molécules à visée thérapeutique( ou encore la nécessité de nouveaux motifs chimiques) s’est tourné vers la préparation d’une petite quantité d’un grand nombre de molécules appelé banque. La mise au point des banques repose sur un mode de synthèse appelé de manière générale : Chimie Combinatoire. Par cette procédure plusieurs millions de molécules peuvent être générés rapidement. Depuis une dizaine d’années, cette technique connaît une croissance importante, d’une part avec la nécessité toujours plus importante de mettre au point de nouvelles molécules pour la synthèse de nouveaux composés bio-actifs, mais aussi par le développement de nouvelles techniques de synthèse. La première méthode développée pour la chimie combinatoire est la synthèse parallèle. Cette méthode consiste synthétiser en parallèle plusieurs composés. Ce type de synthèse s’effectue en général dans des plaques de 96 puits et peut aussi bien être réalisé en phase homogène qu’en phase solide. La stratégie « split and mix » est une autre technique de chimie combinatoire qui est indissociable de la synthèse en phase solide. Ce mode de synthèse permet de générer rapidement un grand nombre de molécule. Le principal inconvénient de la méthodologie « split and mix » est que le mélange répété des billes de résines pose un problème de traçabilité des différents composés. La chimie combinatoire repose sur deux méthodologies de synthèse chacune apportant une spécificité selon le but recherché. Les synthèses « split and mix » permettent l’obtention de banques à diversités maximales alors que la synthèse parallèle est plus adaptée à l’obtention de banques focalisées. Les deux stratégies utilisées en chimie combinatoire diffèrent selon le but recherché mais diffèrent aussi dans leur mode d’exécution.
5.2 Les stratégies utilisées en Chimie Combinatoire
La mise au point de banques de molécules est basée sur deux concepts de synthèse distincts : la synthèse dite « split and mix » et la synthèse dite parallèle. La synthèse parallèle en phase homogène est actuellement la plus utilisée de par la simplicité de mise en œuvre. La synthèse en parallèle en phase solide est quant à elle également très utilisée pour la mise au point de banques focalisées dans le cas de l’optimisation d’une structure. La méthodologie « split and mix » réservée à la synthèse en phase solide, permet l’obtention d’un grand nombre de molécules avec un minimum d’étapes. Lors de ce type de synthèse chaque bille de résines port une molécule individuelle. Cette méthode est plus complexe que la synthèse en parallèle pour la traçabilité des molécules synthétisées bien qu’il existe des méthodes de dé-convolution de banques[36]. La chimie combinatoire utilise donc ces différents modes de synthèse soit pour développer des banques à grande diversité lors de la recherche de nouvelles molécules contre une cible déterminée, soit pour optimiser la structure de molécules déjà connues. Nous allons maintenant discuter les deux concepts de la chimie combinatoire en terme de mise en œuvre et en fonction du nombre de molécules produites.
5.2.1 Synthèse parallèle
La synthèse parallèle a été mise au point dans le milieu des années 80 par Mario Geysen[37] pour la synthèse de peptide. Dans l’exemple ci-dessous une série d’alcools (An) est estérifiée par une série d’halogénures d’acides (Hn) conduisant à neuf esters (figure 3). Dans ce cas, le nombre de nouvelles molécules augmente de façon linéaire avec le nombre de réactions effectuées, la seule limitation en terme de diversité moléculaire est le nombre de synthons disponibles commercialement ou non, ainsi que la diversité de leurs chaînes latérales. En même temps que ces nouvelles méthodologies de synthèse, les chimistes ont également importé de nouvelles techniques telles que les plaques 96 puits empruntées aux biologistes. La synthèse parallèle est parfaitement adaptable au format 96 puits et donc parfaitement automatisable. Si l’on reprend l’exemple ci-dessous en augmentant le nombre d’alcools à 12 et les halogénures d’acides à 8, on obtient rapidement 96 esters qui peuvent ensuite être testés in vitro contre une cible biologique déterminée, ou alors contre un ensemble de cibles.
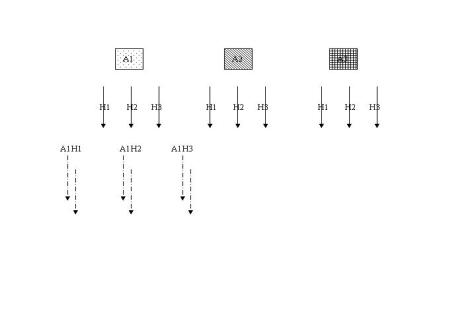
Figure 3 : Représentation de la synthèse en parallèle.
De part la simplicité du processus, la synthèse en parallèle est facilement automatisable et peut s’effectuer soit en chimie en phase homogène, soit en synthèse en phase solide tout en respectant les limitations de chaque mode. Une autre caractéristique de la synthèse en parallèle est qu’une seule molécule à la fois est testée vis à vis de la cible biologique.
5.2.2 Synthèse « Split and Mix »
La méthodologie « spit and mix » a été mise au point à la fin des année 80 par Arpad Furka[38]. Cette technique, repose sur un mélange permanent des différents intermédiaires de synthèse. La synthèse de type « Split and Mix » est utilisée uniquement en phase solide pour des raisons évidentes de manipulation des différents mélanges. Reprenons le cas de trois alcools (An) ancrés séparément sur une résine (figure 4). Les trois alcools ancrés sur le support solide sont alors mélangés, puis de nouveau séparés. Chaque tiers du mélange contenant alors les trois alcools est mis en réaction avec un halogénure d’acide (Hn). Après réaction, les différentes fractions sont une nouvelle fois mélangées. Puis on procède à une nouvelle séparation. Ce protocole est appliqué tout au long du schéma réactionnel.
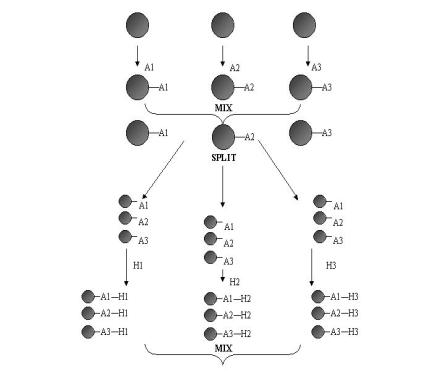
Figure 4 : Représentation schématique de la méthodologie Split and Mix
Dans le cas d’une synthèse « split and mix », le nombre de molécules augmente de façon exponentielle avec le nombre d’étapes effectuées. En effet dans l’exemple ci-dessus un groupe de billes est mis en réaction avec un halogénure d’alkyle et conduit à trois nouvelles molécules. Là encore l’utilisation des plaques 96 puits a permis une automatisation rapide des synthèses. Dans le cas de la méthodologie Split and Mix, un puit représente un ensemble de billes qui portent chacune une molécule individuelle. Lors des tests in vitro pour déterminer si une molécule est active ou non, c’est une moyenne d’activité qui est mesurée et non l’activité d’une molécule individuelle. La méthodologie « Split and Mix » requiert donc deux niveaux de tests pour déterminer quelle est la molécule active dans le mélange. Toutefois si tous les lots présentent une activité insuffisante, la détermination de la molécule active n’est pas poursuivie. Une autre contrainte vis à vis de la méthodologie Split and Mix est que toutes les molécules du mélange réactionnel n’ont pas toutes la même réactivité, et les mélanges ne contiennent donc pas la même proportion des différentes molécules. En conséquence, l’activité potentielle d’une molécule présente dans ce genre de mélange se trouvant en petite quantité peut se trouver « étouffée » par les molécules présentes en grande quantité.
Avec le développement de ces deux méthodologies, l’industrie pharmaceutique a fait un effort considérable pour mettre au point des voies de synthèses automatisées en utilisant de plus en plus de robots et le nouveau défi de ces dernières années est la mise au point de nouveaux modes d’analyses, rapides et précis possédant l’avantage d’être automatisables.
5.3 Comparaison de la synthèse « Split and Mix » et de la synthèse parallèle
Comme nous l’avons vu dans le paragraphe précédent, dans le cas de la synthèse « Split and Mix » chaque puit ne contient pas une molécule unique mais un ensemble de molécules, chaque molécule se trouvant sur une bille de résine. De part cette caractéristique, la méthode Split and Mix requiert deux niveaux de test. Il faut dans un premier temps déterminer quel est le mélange actif et ensuite isoler la molécule active dans le mélange. Il est donc très important de disposer de techniques suffisamment sensibles pour pouvoir déterminer la structure des molécules ancrées sur les différentes billes tout au long de la synthèse, pour mettre en évidence toutes réactions secondaires éventuelles. Dans le cas de la synthèse en parallèle, la molécule active est directement isolée, il n’y a donc pas de problème de dé-convolution comme dans le cas précédent. Un autre inconvénient de la génération de molécules en mélange, est que lors des tests contre les cibles éventuelles, il peut se produire un effet synergique. Un mélange de molécules peut conduire à une détection d’activité mais finalement, lors de la déconvolution aucune molécule n’est trouvée active. Dans le cas de la synthèse parallèle, les molécules étant testées individuellement, il n’existe pas de risque d’effet synergique et donc pas de faux positifs. En ce qui concerne la diversité moléculaire, l’avantage se trouve plutôt du côté de la méthodologie Split and Mix. Au fur et à mesure que le nombre d’étapes augmente, la diversité moléculaire augmente dans le même temps, de par la génération d’un mélange de molécules. Dans les exemples ci-dessus (Figure 3 et Figure 4 ) après le traitement d’un puit, on obtient 3 molécules différentes chacune ancrée sur une bille alors que dans le cas de la synthèse en parallèle on obtient seulement une nouvelle molécule. Les différences mentionnées au cours de ce paragraphe sont inhérentes à la méthodologie utilisée, mais de nouvelles différences s’ajoutent en fonction du mode de synthèse utilisé (synthèse en phase solide/homogène). Ces différences liées au mode de synthèse utilisé seront abordées au cours des paragraphes suivants.
5.3.1 Obtention de banque de molécules de manière ordonnée
Dans le cas de la synthèse en parallèle, il est très facile de suivre où se trouve exactement chaque molécule (les réactions étant effectuées séparément). En vue d’une automatisation, des logiciels peuvent parfaitement donner la traçabilité des molécules en fonction de leurs coordonnées XY sur les plaques 96 puits.
En ce qui concerne la stratégie Split and Mix, il est plus difficile de savoir précisément où se trouve un composé donné puisque après chaque étape, les billes sont mélangées. Pour assurer un suivi de chaque molécule, l’expérimentateur doit recourir à des stratagèmes pour suivre pas à pas un composé. Les techniques utilisées consistent à effectuer un étiquetage soit des molécules incorporées, soit du support solide. L’étiquetage des molécules se fait souvent en utilisant un marqueur qui devient fluorescent en présence d’une autre entité. En ce qui concerne l’étiquetage du support solide, la tâche est plus délicate et requiert souvent l’incorporation d’un émetteur de radiofréquence qui permet de suivre le support en tous moments de la synthèse. L’utilisation d’un émetteur de radiofréquence pour la synthèse automatique n’est pas compatible avec l’utilisation des résines conventionnelles. Pour faire face à ce nouveau problème de la synthèse en phase solide, il a fallu développer de nouveaux supports de synthèse tels que les PINS[39], les lanternes[40] ou bien encore une encapsulation de résines conventionnelles dans une matrice de polymères[41], ces derniers supports permettant l’incorporation aisée d’un émetteur de radiofréquence. Vaino et al.[42] ont mis au point un type de support à forme déterminée. La forme ou encore la couleur de ces supports permet de suivre à tout moment d’une synthèse le devenir d’une molécule. Ces nouveaux supports macroscopiques sont caractérisés par des charges élevées (de l’ordre de la dizaine de mmole), donnant une quantité suffisante de molécules pour plusieurs tests biologiques après coupure, mais pouvant également permettre une analyse directe de la molécule ancrée sur le support à condition de disposer d’une technique d’analyse adaptée. L’analyse directe d’une réaction chimique sur une résine utilisée classiquement en synthèse organique en phase solide n’est pas adaptée à l’utilisation de robots, de plus en plus utilisés pour la chimie combinatoire pour des raisons pratiques évidentes. En effet, le prélèvement de résine dans le milieu de synthèse n’est pas facile à mettre en oeuvre. Les nouveaux supports de synthèse dont nous venons de parler sont particulièrement adaptables à la chimie combinatoire, en particulier dans le cas de la synthèse « Split and Mix ». Il est donc très important de développer des méthodes de suivi de réaction sur ce type de support, possédant une fonctionnalité de surface.
5.3.2 Adaptabilité des réactions pour obtenir des bibliothèques combinatoires
Adapter une réaction chimique dans le but d’obtenir un ensemble de molécules de façon combinatoire est un problème qui n’est pas insurmontable pour la synthèse en phase homogène. En théorie au moins, les problèmes rencontrés peuvent être résolus en modifiant des paramètres tels que la température, le solvant ou encore la stœchiométrie de la réaction.
Par contre, dans le cas de la synthèse en phase solide, il est beaucoup plus difficile de surmonter ces problèmes notamment à cause de la nécessite d’un point d’ancrage. En effet, pour adapter une réaction de façon standard, différents problèmes doivent être résolus. Tout d’abord il faut trouver le « linker » approprié, avec une méthodologie de coupure simple. Un autre problème découle directement du point d’ancrage. En effet, toutes les molécules à synthétiser doivent posséder une fonction permettant l’ancrage sur le support solide. Cette fonction chimique est souvent un alcool, une amine, ou encore un acide mais cette fonctionnalité n’est pas toujours compatible avec la molécule finale. Pour surmonter cette difficulté, de nombreux efforts doivent être investis pour développer de nouveaux « linker » dits « sans trace » (traceless linker), ou encore il faut développer de nouvelles méthodologies de clivage qui consistent par exemple à former un cycle lors de la séparation de la molécule du support solide. Tous les efforts investis pour appliquer un protocole standard sur la phase solide ne sont pas toujours compatibles avec le besoin croissant de nouvelles molécules pour l’industrie pharmaceutique.
6 Conclusion
Dans ce chapitre nous avons vu que la synthèse en phase solide a contribué à l’avancement de la synthèse de peptides et d’oligonucléotides, notamment avec la possibilité d’automatisation des synthèses, du fait de la simplicité de la chimie utilisée. La synthèse en phase solide a également contribué au développement de la chimie combinatoire. Malgré les avancées réalisées, ce mode de synthèse soulève de nouvelles questions. Dans le cas de la synthèse peptidique par exemple, quels sont les facteurs qui limitent la synthèse de certaines séquences, la structuration d’un peptide est-elle possible lorsque celui-ci est encore ancré sur le support solide ? Nous avons vu qu’un des grands inconvénients de la chimie en phase solide est le manque de techniques d’analyses permettant une identification et une quantification directe des molécules ancrées sur le support solide. Ce manque de techniques d’analyses est devenu encore plus important avec le développement de la chimie combinatoire, car lors de l’optimisation d’une réaction pour le développement d’une banque, il est primordial de pouvoir suivre la réaction sur le support afin de pouvoir détecter les produits secondaires éventuels le plus tôt possible. Ma thèse traitera ces différents problèmes dans le cadre du développement de la RMN HRMAS. L’identification et la quantification des produits ancrés sur des supports solides seront décrites, ainsi que la détection rapide d’impuretés. Dans le troisième chapitre nous étudierons la structuration de peptides sur différentes résines par la même technique. Le dernier chapitre sera consacré à un développement très récent de la technique HRMAS pour l’étude de molécules dans le contexte cellulaire.