INTRODUCTION......................................................................................................................................................................
I ) BIBLIOGRAPHIE.................................................................................................................................................................
II) Travaux personnels................................................................................................................................................
1) Synthèse de -Na-(t-butoxycarbonyl)-L-alaninal..................................................................................
2) Synthèse de l’alcène cétal et de son hydrolyse...............................................................................
2-1 Réaction de Wittig..................................................................................................................................................
2-2 Essai d’hydrolyse de la fonction acétal
du composé 13..................................................................................
3) Synthèse de 15 par
condensation de WITTIG avec le bromure de triphenyl
(3-triméthylsilylprop-2-ynyl) phosphonium et oxydation en acide..............................................................................................................
3-1 Réaction de Wittig..................................................................................................................................................
3-2 Hydroboration oxydante de
N-(t-butoxycarbonyl)-5(S)-amino héx-3-ène-1-yne-1trimethylsillyle.......
4) Synthèse de 18 et oxydation en acide........................................................................................................
4-1 Synthèse du sel de phosphonium.........................................................................................................................
4-2 Condensation de Wittig.........................................................................................................................................
4-3 Oxydation directe de 18.......................................................................................................................................
III) Partie expérimentale...........................................................................................................................................
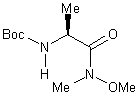
1) -N-a-(t-butoxycarbonyl)-N-méthyl-N-méthoxy
L-alaninamide 10.............................................
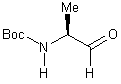
2) -N-a-(t-butoxycarbonyl)-L-alaninal
11......................................................................................................
3) N-(t-butoxycarbonyl)-5(S)-amino hex-3-ènal propylène cétal (E) 13.......................................
4) Synthèse de N-(t-butoxycarbonyl)-5(S)-amino
hex-3-ène-1-yne-1-triméthylsillyle 15..
5) Synthèse de (S)-N-(t-butoxycarbonyl)-5-aminohex-3-ènoique acide
(E) 17............................
6) 3-tosyloxypropanol 20........................................................................................................................................
7) éther tétrahydropyrannique-3-tosyloxypropanol 21..................................................................
8) Ether tetrahydropyrannique -3-iodopropanol 22............................................................................
9) Iodure propanol triphényl phosphonium 23..........................................................................................
10) Synthèse de l’iodure de l’éther tetrahydropyrann de propyl
triphényl phosphonium 19
11) Synthèse de N-(t-butoxycarbonyl)-5(S)-amino hex-3-ènal
tetrahydropyranyl (E) 18
12) Synthèse de (S)-N-(t-butoxycarbonyl)-5-aminohex-3-ènoique acide
(E) 17..........................
Références bibliographique..................................................................................................................................
INTRODUCTION
La modulation de la réponse
immunitaire, constitue une thérapeutique de choix dans le traitement des
maladies auto-immunes ou de la prévention du rejet des greffes.
Cette réponse immunitaire est un phénomène très complexe
dont une des étapes est la fixation de la protéine antigénique sur un récepteur
des lymphocytes T appelé TcR. Une des molécules responsable de cette fixation
est le CMH (Complexe Majeur d’Histocompatibilité) qui se lie avec des fragments
peptidiques de l’antigène, ayant subit au préalable, une protéolyse partielle.
Il est donc important de connaître la nature et quels
sont les résidus concernés dans la liaison entre le CMH et les parties
peptidiques. Ainsi la mise en évidence des requis structuraux nécessaire à un
peptide pour se lier au CMH et l’identification des chaînes latérales,
indispensables à une reconnaissance par le TcR, permettraient la mise au point
de pseudopeptides « compétiteurs » utilisables pour le traitement des
maladies auto-immunes. Un fragment HEL du lysozyme de blanc d’œuf est un déterminant T bien étudié, dont la
séquence antigénique est bien connue. Ce travail est donc une contribution à la
détermination du rôle du squelette peptidique de cette molécule.
Pour cela nous avons prévu
la synthèse d’isostère éthylénique de la liaison amide :
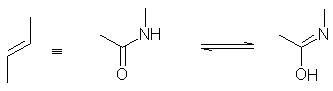
Les pseudopeptides synthétisés seront alors incorporés dans le
décapeptide HEL (52-61), à la place des acides aminés correspondants.
I) BIBLIOGRAPHIE
I ) BIBLIOGRAPHIE
Les premières
synthèses de pseudopeptides éthyléniques remontent au début des années 80, en
particulier avec le pseudo-dipeptide Tyry(CH=CH)Gly [1].
De nombreuses raisons
justifient le remplacement de la liaison amide par une double liaison. D’une
part la liaison éthylénique offre une meilleure résistance aux dégradations
enzymatiques (amidases), elle est plus lipophile et offre ainsi une meilleure diffusion
à travers la paroi cellulaire. Le remplacement par une liaison éthylénique va
permettre après incorporation dans le décapeptide d’explorer la restriction
conformationnelle et le rôle des liaisons hydrogènes puisque en remplaçant la
liaison amide par une oléfine on supprime celles-ci. D’autre part le choix
d’une liaison éthylénique en remplacement d’une liaison amide se justifie au
point de vue des angles et de la longueur des liaisons [1] :

La liaison amide étant en conformation transoïde, l’oléfine isostère
devra être en configuration E.
Ils existent plusieurs méthodes de synthèse de pseudopeptides
éthyléniques mais nous nous sommes intéressés qu’aux méthodes stéréosélectives
au niveau de la double liaison.
Synthèse par oléfination de Julia [2]
Une double liaison
isostère d’amide peut être obtenue par
réaction entre un aldéhyde et une sulfone. Cette méthode est la première a
avoir été décrite qui permette d’obtenir de façon stéréoseléctive des
pseudodipeptides éthyléniques comportant coté C-terminal un autre acide aminé
que la glycine.
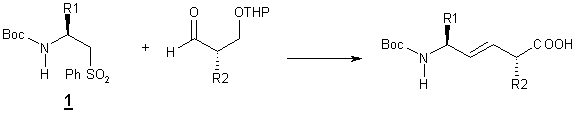
(R1= CH2PhOH, CH2Ph, CH2CH(CH3)2)
La sulfone 1 est
obtenue en quatre étapes à partir de l’aminoacide correspondant. La double
liaison est obtenue avec une géométrie trans.
Synthèse par alkylation de Trost
Le dipeptide
isostère Tyr-Y[CH=CH]-Gly a été synthétisé par Thompson [3] et collaborateurs à
partir N-Boc-tyrosinal 2 qui est transformé en acétate
allylique 3.
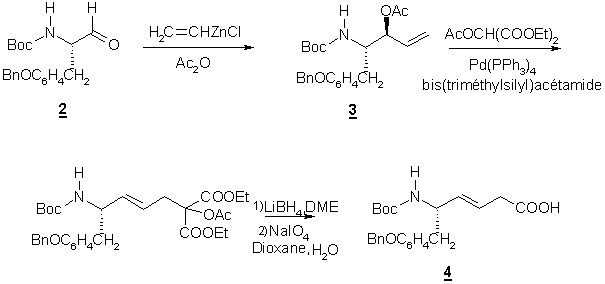
Lors de cette synthèse les auteurs ont obtenu le dipeptide 4 avec un rendement de 28 % à
partir de l’aldéhyde 2.
Synthèse par cycloaddition [4+2] et
réarrangement de Claisen
Whitesel et Lawrence
[4] décrivent en 1989 la synthèse de différents dipeptides isostères contenant
une double liaison trans. Ces
composés sont obtenus de façon séréoselective par cycloaddition [4+2] suivie
d’un réarrangement de Claisen. Le dipeptide D-Ala-Y[CH=CH]-Ala est obtenu avec un rendement totale de 20 % à partir du N-sulfinylcarbamate du 8-phényl menthol 5 et du trans-trans
-2,4-hexadiène.
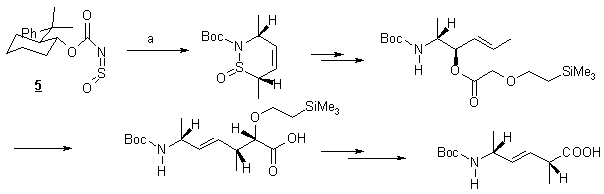
(a : trans-trans-2,4-hexadiène, SnCl4)
Synthèse à partir d’esters a,b-insaturés g-oxygénés
La synthèse stéréosélective de nombreux dipeptides du type AA1-Y[CH=CH]-AA2 a été étudié par Ibuka et coll. [5] en utilisant
des esters a,b-insaturés g-oxygénés synthétisés 7
à partir d’acides aminés N-a-protéges 6.
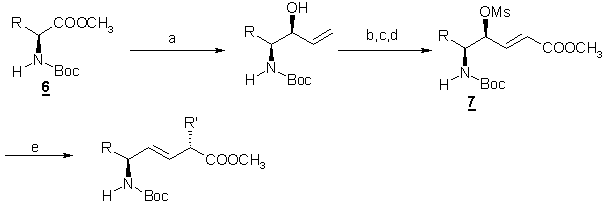
(a : i DIBAL, ii ClMgCH=CH2 ; b : DMAP, MsCl,
pyr ; c : O3, -78°C ; PPh3=CHCO2CH3,
-78°C ; d : CH3SO2Cl, -78°C ; e :
RCu(CN)Li_BF3)
Synthèse
basée sur une condensation de Wittig
Les synthèses basées
sur des condensation de Wittig utilisent des sels de phosphonium possédant un
équivalent de la fonction acide soit sous forme de cétals soit sous forme
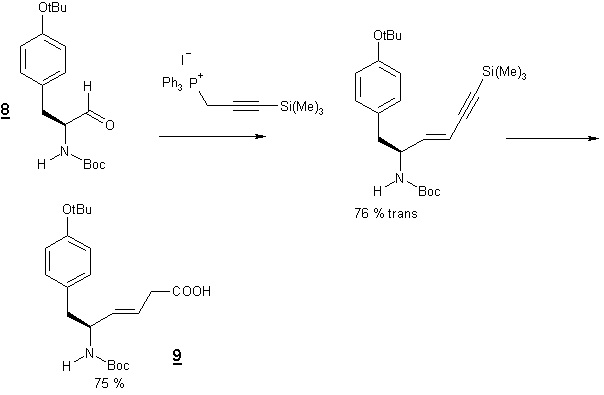
d’éther sillylé. Hann et collaborateurs [6] ont décrit une synthèse du
pseudopeptide Tyr-y-(CH=CH)-Gly 9 à
partir de la N-Boc-Tyrosine 8
qui après réduction est couplé avec le bromure de (3-triméthylsilyl
prop-2-ynyl) triphényl phosphonium. Cette condensation de Wittig donne le
dérivé trans avec 76 % de rendement.
L’oxydation conserve la stéréochimie de la double liaison.
Une seconde voie d’accès au dipeptide éthylénique Tyry(CH=CH)Gly est décrite par Hann
et collaborateurs [6], dans laquelle le substrat de départ est toujours la
tyrosine protégée mais par contre le sel de phosphonium utilisé est le chlorure
de (2-carboxyéthyl) triphénylphosphonium. L’inconvénient de cette méthode est
son faible rendement et l’obtention de deux isomères cis-trans dans la proportion 50/50.
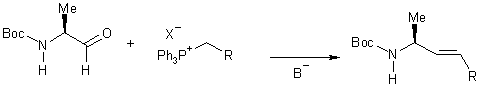
Compte tenu des résultats de la littérature
concernant la synthèse de pseudopeptides éthyléniques, la synthèse que nous
proposons repose sur une condensation de WITTIG entre l’alaninal dont la
fonction amine est protégée sous forme de carbamate par un groupement
terbutoxycarbonyl (Boc) et un sel de phosphonium correctement substitué afin
d’obtenir ultérieurement la fonction acide correspondant à la glycine.
La fonction acide de la partie glycine pourra être obtenu par
différentes stratégies.
-
oxydation d’une fonction alcool, préalablement protégée ou non sous forme
d’éther tétrahydropyranyle [7] :
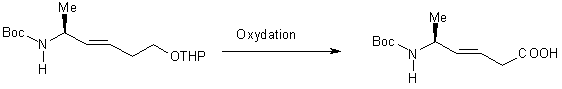
- oxydation d’une fonction aldéhyde préalablement protégée sous forme
d’acétal [8] :

- à partir d’un alcyne par hydroboration et oxydation de l’organoborane
intermédiaire [9] :
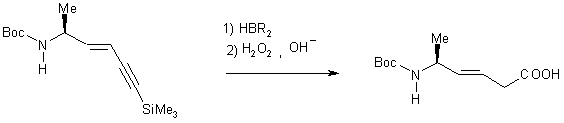
Ces trois stratégies seront étudies afin de trouver la meilleure voie
de synthèse. Les résultats obtenus lors des différents essais effectués selon
ces trois méthodes sont présentés dans la partie travaux personnels.
II) TRAVAUX PERSONNELS
II) TRAVAUX
PERSONNELS
Le but de ce projet est la
synthèse d’un peptide isostère éthylénique Alay(CH=CH)Gly.
Le substrat de départ des
différentes voies de synthèse est l’alanine dont la fonction amine est protégée par un groupement terbutoxycarbonyl
(Boc) .
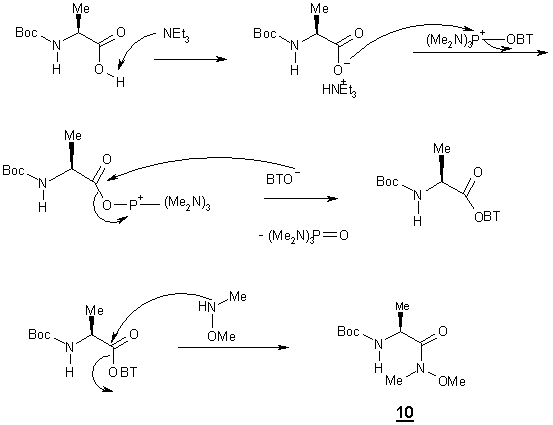
1) Synthèse de -Na-(t-butoxycarbonyl)-L-alaninal
Le Na-(Boc) alaninal 11 est synthétisé en deux étapes à partir de la N-Boc alanine, par
réduction de l’amide de Weinreb 10
selon la méthode de Fehrentz et Castro [10]. Afin d’obtenir cet amide la fonction acide est activé par le groupement
BOP (benzotriazolyl-oxy-tris-(di-méthylamino)-phosphonium hexafluorophosphate),
puis cet intermédiaire réagit avec la N-méthoxy-N méthyle amine pour conduire à
l’amide 10 avec un excellent
rendement de 90%.
La seconde étape est la réduction 10 obtenu précédemment selon le protocole de Fehrentz et
Castro [10].
Cette réduction s’effectue à
0°C à l’aide de deux équivalents de LiAlH4. Cette réaction conduit à
deux produits, dont l’un est très
largement majoritaire et qui correspond à l’aldéhyde 11 attendu avec un rendement de 80 %, la RMN du proton
montre l’apparition d’un proton aldéhydique à 9,8 ppm, et la spectroscopie IR montre une nouvelle
bande carbonyle à 1733 cm-1).
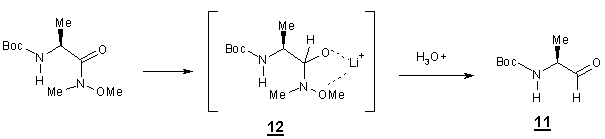
La réaction s’arrête au
stade de l’aldéhyde 11 car il
existe un état de transition 12
ou le lithium est fortement chélaté par
l’oxygène et l’azote, conduisant ainsi après hydrolyse à l’aldéhyde. Le second
produit dont nous n’avons pas pu déterminé la structure ( ce n’est pas l’alcool
absence de bande à 3400 cm-1 en IR) représente 5 % en masse du
mélange. Afin d’obtenir une plus grande quantité de produit secondaire pour
déterminer sa structure nous avons modifié les conditions opératoires. Nous
avons constaté que si le temps de réaction était augmenté, le rendement de
réaction diminuait.
Tableau 1
Evolution du rendement de formation de 11 en
fonction du temps
|
Temps de réaction (h) |
Rendement (%) |
|
0,5 |
80 |
|
1 |
68 |
|
1,5 |
60 |
|
4 |
51 |
Malheureusement nous n’avons
pas réussi à déterminer la structure du produit secondaire, par spectroscopie
Infra Rouge nous pouvons dire qu’ il ne s’agit pas de l’alcool.
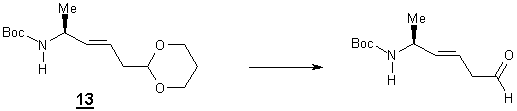
2)
Synthèse de l’alcène acétal 13 et essais de son
hydrolyse
2-1
Réaction de Wittig
Cette condensation se déroule à basse température
(-78°C), la base utilisée est l’hexaméthyldisillylamidure de potassium (KHMDS).
Lors de cette synthèse l’alcène est obtenue avec 83 % de rendement. L’analyse
RMN montre la présence des protons éthyléniques (d=5,38 ppm, d=5,47 ppm ) dont la constante de couplage de
18 Hz indique la stéréochimie trans.
2-2 Essai
d’hydrolyse de la fonction acétal du composé 13
Afin
d’hydrolyser la fonction acétal, nous avons essayé différents modes
opératoires.
- Tout d’abord le produit à été
traité dans l’acétone à reflux avec une quantité catalytique d’acide
paratoluènesulfonique [11].
Après 6h de réaction on
obtient deux produits en faible quantité et une grande quantitée de produit de
départ (65 %). Nous n’avons pas pu déterminer la structure des deux produits
minoritaires, mais la RMN du proton n’à pas montré la présence de la fonction
aldéhydique. Aussi , nous avons testé d’autres conditions opératoires telles
que:
-l’acétate d’éthyle saturé en eau avec une
quantité catalytique de DDQ [12]
-tétrabromométhane avec de la
triphénylphosphine dans le dichlorométhane[13]
-FeCl3, 6H2O dans le
dichlorométhane [14]
-CeCl3, 7H20 dans le dichlorométhane [15]
Aucune de ces réactions n’a conduit à l’aldéhyde,
seules les réaction avec le sel de fer et celle avec le sel de cérium ont
montré une disparition du produit de départ, un nombre important de composés
sont détectés en CCM.
D’après Baudin [16], les acétals cycliques sont plus
stables et plus difficiles à hydrolyser que les diéthylacétales. Aussi nous
avons pensé réaliser une transacétalisation en un composé 14 qui serait ultérieurement hydrolysé plus facilement en
aldéhyde. Le traitement de 13
dans l’éthanol en présence d’une quantité catalytique d’acide
paratoluènesulfonique [17] nous à conduit à 14 avec un rendement de 22 % seulement, après 24 heures à
température de reflux du solvant.
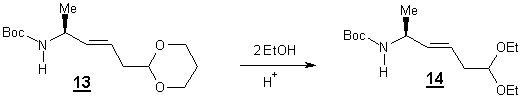
Malheureusement ,
l’hydrolyse de 14 n’a pas
donné l’aldéhyde attendu, le produit se dégradant dans ces conditions
opératoires.
L’oxydation
directe du cétal 13 en acide
par le réactif de Jones [8] a une fois encore conduit à un mélange complexe de
produits duquel nous n’avons pu isoler le produit recherché.
Ces résultats peuvent
s’expliquer selon les conditions utilisées soit par une faible réactivité du
cétal (le produit de départ est alors récupéré en fin de réaction), soit par la
faible stabilité de l’aldéhyde (on récupère alors un mélange complexe de produits
en fin de réaction). Dans le cas de l’oxydation des réactions secondaires
peuvent avoir lieu, telle que l’hydrolyse du groupement Boc en milieu très
acide (réactif de Jones). Ainsi nous nous sommes orienté vers une autre
stratégie.
3)
Synthèse de 15 par condensation de WITTIG avec le bromure de triphenyl
(3-triméthylsilylprop-2-ynyl) phosphonium et son oxydation
en acide
3-1 Réaction de Wittig
L’étape de
condensation de WITTIG est l’étape déterminante de la synthèse car c’est lors
de cette étape qu’est fixée la stéréochimie de la double liaison.
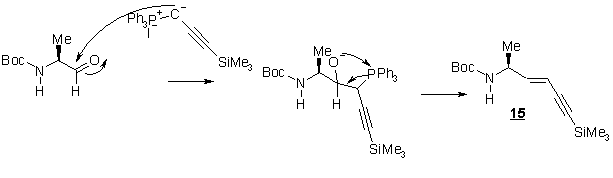
La base utilisée étant le nBuLi cette réaction est
réalisée à basse température (-78°C) Un
mélange de deux produits est obtenu, le produit majoritaire est isolé avec 77 %
de rendement. Les analyses IR et 1H RMN ont permis de caractériser
celui-ci : d’une part la spectroscopie IR montre la disparition de
la bande C=O correspondant à l’aldéhyde, et d’autre part par la RMN du proton montre l’apparition du pic
correspondant au groupement SiMe3 (0,2 ppm). La mesure de
la constante de couplage (J=18 HZ) permet de conclure à la stéréochimie trans
de la double liaison du produit. Le produit minoritaire est l’isomère cis (les deux protons éthyléniques
résonnent à d 5,52 ppm sous forme d’un
doublet (J=11 Hz) et d 5,86 ppm sous forme d’un
massif complexe.
3-2
Hydroboration oxydante de N-(t-butoxycarbonyl)-5(S)-amino
héx-3-ène-1-yne-1trimethylsillyle
L’alcyne 15 est dans un premier temps traité par le dicyclohexylborane en excès préparé extemporanément. L’organoborane intermédiaire 16 est oxydé en milieu basique par l’eau oxygénée.

Les conditions opératoires
ont été difficile à mettre au point. Les conditions utilisées par Hann [6] ne
nous ont pas permis d’isoler facilement l’acide 17. En effet nous avons obtenu des quantités importantes de
cyclohexanol, formées par hydrolyse de l’organoborane intermédiaire et
difficile à éliminer complètement. D’autre part les lavages acides ont conduit
à l’hydrolyse du groupe protecteur Boc. Après de nombreux essais d’extraction,
les meilleurs résultats ont été obtenu en utilisant un tampon acide
citrique/citrate à pH 3. Dans ces conditions l’acide 17 a été obtenu avec un rendement de 56 %. Nous l’avons
caractérisé en spectroscopie IR bande d’absorption à 3450 cm-1 nOH acide et en RMN du carbone treize pic à 160 ppm
C=O acide).
Les difficultés de synthèse (rendement moyen) et le coût des réactifs en particulier l’alcyne sillylé, nous ont conduit à envisager la synthèse d’un autre sel de phosphonium par une voie utilisant des réactifs moins onéreux.
4) Synthèse de 18 et
oxydation en acide
Nous avons envisagé d’obtenir 18 par condensation de Wittig entre l’aldéhyde 11
et le sel de phosphonium 19.

19 est synthétisé en cinq étapes
4-1
Synthèse du sel de phosphonium
19
Le
propan-1,3-diol est monotosylé par le chlorure de tosyle en défaut en présence de triéthylamine. La
monotosylation s’effectue avec un rendement de 90 % par rapport au chlorure de
tosyl.

La spectroscopie Infra Rouge
montre l’apparition des bandes caractéristiques des aromatiques et de SO2 (3080
cm-1 nCH aromatique 1326 cm-1 SO2).
La fonction alcoolique de 20 est alors transformée en
éther tetrahydropyrannique par le DHP en présence d’acide
paratoluènesulfonique.

Le produit 21 est obtenu en 30 min avec 95
% de rendement, l’analyse IR confirme la protection de l’hydroxyle par la
disparition de la bande d’absorption de l’hydroxyle (3350 cm-1).
Dans un troisième temps le
groupement tosylate est substitué par l’iodure par la réaction de Finkelstein
[18].
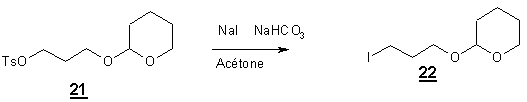
La substitution du tosylate
est obtenue après 4h d’agitation de 21
en présence de NaI à température ambiante dans l’acétone avec un rendement de
95 %. En spectroscopie infra rouge on constate la disparition des bandes
aromatiques, et l’apparition d’un bande à 550 cm-1 correspondant à
la liaison C-I.
Pour l’obtention de 24
il fallait substituer l’iodure par la triphénylphosphine. Cette réaction est
réalisée dans le mésitylène à reflux, après une heure d’agitation à 170°C, un
produit précipite. Les analyses IR et RMN ont montré que le produit obtenu
était en faite le sel de phosphonium dont la fonction éther
tetrahydropyrannique était hydrolysée (apparition d’une bande d’absorption à 3400 cm-1 ). 23 est obtenue avec un rendement
80 %. Le traitement de 22 en
absence de triphénylphosphine à reflux du mesitylène conduit à l’alcool
correspondant avec une rendement de 73 %. Cette réaction montre que l’hydrolyse
pourrait être du à la présence d’eau dans le solvant (qui n’a pas été distillé
avant usage). Ainsi le produit 23
a été reprotégé.
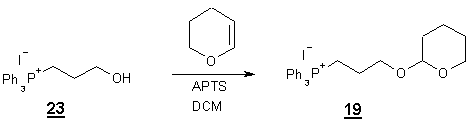
19 a été obtenu avec 80 % de
rendement.
4-2
Condensation de Wittig
Le première essai de
condensation a été effectue à -78°C en utilisant KHMDS comme base, la CCM a
montré que la condensation ne s’était pas produite ; en effet on ne
constate pas la disparition de l’aldéhyde 11
qui est récupéré intégralement à la fin de la réaction.

Avec le BuLi nous avons mis
au point les meilleurs conditions expérimentales (temps d’agitation, rapport
entre les réactifs) afin d’obtenir le meilleur rendement. Après de multiples
essais le meilleur rendement en 18
était de 60 %. La RMN du proton ne nous à pas permis de déduire la stéréochimie
exacte de la double liaison car les protons éthyléniques résonnent sous forme
de massifs complexes.
4-3
Oxydation directe de 18
Nous avons effectué un essai
d’oxydation directe de l’éther tétrahydropyrannique par le réactif de Jones[8] dans les conditions de Hanessian et
coll. pour obtenir . Après purification , nous obtenons le même rf en CCM et le
même spectre infra rouge que celui obtenu dans le paragraphe 3-2.
Cette méthode à la
différence des précédentes, présente l’avantage d’être peu coûteuse compte tenu
des réactifs utilisés pour la synthèse du sel de phosphonium, malgré tout le
rendement de condensation reste à être améliorer.
Conclusion
Sur les trois voies de
synthèse étudiées deux seulement nous ont permit d’accéder de façons
satisfaisantes au composé cherché. Ces méthodes permettent l’accès à des
pseudodipeptides éthyléniques en quatre étapes à partir de l’acide aminé
protégé. Ces deux voies permettent d’obtenir 5 avec des rendements globaux de 32 et 20 %. Ayant mis au
point la synthèse du pseudopeptide 17
l’objectif de ce projet est de substituer celui-ci de façon stéréosélective en
utilisant la méthode d’Evans [19].
III) PARTIE EXPERIMENTALE
Liste des abréviations
utilisées
AA : acide aminé
AE : acétate d’éthyle
APTS : acide paratoluène sulfonique
Boc : tert-butoxycarbonyle
BOP :
benzotriazolyl-oxy-tris-(di-méthylamino)-phosphonium héxafluorophosphate
BuLi : Butyllithium
DCM : dichlorométhane
DDQ : 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
DMAP : 4-diméthyl amino pyridine
EE : éther éthylique
EP : éther de pétrole
MeOH : méthanol
PPTS : pyridinium paratoluène sulfonyl
pTsOH : acide
paratoluènssulfonique
III) PARTIE EXPERIMENTALE
1) -N-a-(t-butoxycarbonyl)-N-méthyl-N-méthoxy L-alaninamide 10
6,4 mmoles (0,9 ml) de
triéthylamine et 6,4 mmoles (2,9 g) de BOP sont ajoutées en une seule fois à
une solution de N-BOC alanine 6,4 mmoles (1,2g) dans 25 ml de dichlorométhane.
Après 5 min d’agitation à
température ambiante 7,6 mmoles (0,74g)
de chlorhydrate de N-méthoxy-N-méthylamine et à nouveau 7,6 mmoles (1ml) de triéthylamine sont
ajoutés.
Le mélange est agité à
température ambiante pendant 1h30. La solution est reprise avec 25 ml de
dichlorométhane. La phase organique est lavée avec une solution de HCl (10%)
(3x30ml), une solution saturée d’hydrogénocarbonate de sodium (3x30ml) et enfin
une solution saturée de chlorure de sodium (3x30ml). La phase organique est
séchée sur sulfate de sodium puis évaporée sous vide. Le produit brut est
purifié sur colonne de silice (EE/EP=80/20). 5,76 mmoles d’un solide blanc sont
obtenues.
Rdt=90%
Point de fusion :152°C Litt :150°C [10]
IR cm-1: 3292 (nNH), 2976, 2935 (nCH), 1706 (nC=O amide), 1660 (nC=O BOC), 1185 (dC-O).
1H RMN (CDCl3) (200
MHz) : 1,32 (d, J= 6.8Hz, 3H,
NCHCH3) , 1,45 (s, 9H, (CH3)3C) ,
3,21 (s, 3H, NCH3) , 3,77 (s, 3H, OCH3)
, 4.13 (m, 1H, CH3CH) , 5,20 (s, 1H, NH) ,
2) -N-a-(t-butoxycarbonyl)-L-alaninal 11
Une solution de 3,4 mmoles
(0,8g) de 10 dans 20ml d’un
mélange éther-THF (1/1) est coulée sur
une solution d’hydrure de lithium aluminium 6,1 mmoles (0,23 g) dans 10 ml de
THF à 0°C sous azote.
Le mélange est laissé sous
agitation à 0°C pendant 30 min.
Le milieu est traité à 0°C avec 10 ml d’une solution
d’hydrogénosulfate de sodium (0.35M).
La phase aqueuse est séparée
et extraite par de l’éther éthylique (3x30ml). La phase organique est lavée
successivement avec 3x20ml d’une solution d’HCl (10%), une solution saturée de
NaHCO3 puis une solution saturée de NaCl. La phase éthérée est
séchée sur sulfate de sodium puis évaporée sous vide. On obtient un produit
sous forme d’une pâte blanche qui est purifié sur colonne de silice
(AE/EP=70/30). 2,72 mmoles de cristaux blanc sont obtenus.
Rdt=80%.
Point de fusion 88°C Litt : 88-89°C [10]
IR (cm-1) :
3329 (nNH) ; 2978,2934 (nCH) ; 1733 (nCHO) ; 1681 (nC=O BOC).
1H RMN (CDCl3) (200 MHz) :1,32 (d, J=5,25
Hz , 3H , NCHCH3) ; 1,46 (s, 9H, (CH3)3C) ;
4,21 (m, 1H, NCH) ; 5,1 (m, 1H, NH) ; 9,6 (s, 1H, CHO).
3) N-(t-butoxycarbonyl)-5(S)-amino hex-3-ènal propylène cétal (E) 13

6,2 mmoles (12 ml) d’une solution d’héxaméthyldisilylamidure de
potassium (0,5 M dans du toluène) sont coulées à température ambiante sous
azote sur 6,2 mmoles (2,82 g) du bromure de 2-(1,3-dioxan-2-yl)
éthyl-triphénylphosphonium dissoutes dans 70ml de THF.
Le mélange réactionnel est
laissé sous agitation à température ambiante pendant 1h. 3,1 mmoles (0.53 g)
d’une solution de N-a-(t-butoxycarbonyl)-L-alaninal
11 dissout dans 30ml de THF
sont ajoutées lentement à -78°C.
Le mélange réactionnel est
agité pendant 1h à -78°C puis ramené progressivement à température ambiante.
30 ml de méthanol sont
ensuite ajoutés puis le mélange réactionnel est versé dans 100 ml d’une
solution saturée de tartrate de sodium et de tartrate potassium (1/1). La phase
aqueuse est extraite par de l’éther éthylique ( 3x30ml ). Après évaporation le
produit est purifié sur colonne de silice ( DCM/MeOH 99/1 ). 2,63 mmoles
d’alcène sont obtenues sous forme d’un solide blanc.
Rdt=85%.
Point de fusion : 56°C
IR (cm-1):
3364 (nNH) ; 2972,2931,2852 (nCH) ; 1699 (nC=O BOC) ; 1630 (nC=C) ; 944 (nHC=).
1H RMN (CDCl3) (300 MHz): 1,21 (d,
J=9Hz,3H, NCHCH3) , 1,45 (s, 9H, (CH3)3C) ,
2,05-2,015 (m, 2H, NCHCH3) , 2,45 (t, J=3Hz, 2H, CHCH2CH) ,
3,75 (m, 2H, CH2CH2CH2) , 4,07 (m,
4H, OCH2CH2) , 4,36 (M,1H, NH) , 4,56
(t, J= 6 Hz,1H, CH2CHO) , 5,38 (m, J=10 Hz,1H, CH=CH) ,
5,47 (m, J=16 Hz,1H, CH=CH)
4) Synthèse de N-(t-butoxycarbonyl)-5(S)-amino hex-3-ène-1-yne-1-triméthylsillyle 15
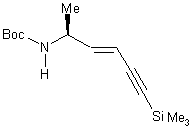
3,2 ml de butyllithium (1,6M) sont coulés sur une
solution de bromure de triphényl(3-triméthylsilylprop-2-ynyl) phosphonium 4,3
mmoles (1,96 mg) dans 10 ml de THF. Le
mélange est agité 30 min à -60°C.
Une solution de N-BOC alaninal 2,8 mmole (500 mg) dans 10ml de THF est
coulée sur la solution précédemment obtenue. A la fin la température est laissée remontée à 15-20°C.
Le mélange est agité 1h à
température ambiante.
Après une heure 15 ml d’une
solution saturée de chlorure de sodium sont ajoutés.
Le mélange est extrait à
l’éther de pétrole, séché sur sulfate de sodium puis évaporé sous vide.
L’oxyde de trphényl
phosphine formé est éliminé par filtration après trituration dans l’éther de
pétrole et le filtrat est évaporé sous vide. 2,6 mmoles d’une huile
translucides sont obtenues, l’huile est
purifiée sur colonne de silice ( AE/EP 10/90 ). 2,1 mmoles de cristaux
blancs sont ainsi obtenus.
Rdt : 77%.
IR (cm-1) :
3338 (nNH), 2975,2931 (nCH), 2155 (nC=C), 1250 (dSi-Me), 850 (gSI-Me)
1H RMN (CDCl3) (300MHz) : 0,18 (Si(CH3)3, 1,20 (d, j =4Hz, 3H, CHCH3), 1,40 (s, 9H, (CH3)3C, 4,20 (m,1H, CHCH3), 4,5 (m, 1H, NH), 5,6 (d, j=14Hz ,1H, CH=), 6,2 (d, j=14 ,1H, CH=).
5) Synthèse de l’acide
(S)-N-(t-butoxycarbonyl)-5-aminohex-3-ènoique (E) 17
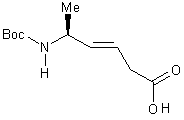
17 ml d’une solution de BH3/THF
sont ajoutés goutte à goutte à 34,3 mmoles de cyclohexène (19 ml) à 0°C.
Le mélange est agité pendant
3 h à 0°C.
Une solution de 15 3,4 mmoles (800 mg) dissout
dans 20ml de THF est coulée lentement
sur la solution précédente.
Le mélange est agité pendant
2 h à température ambiante.
Après 2 h d’agitation 2 ml
de MeOH et 4 ml de NaOH (2N) sont ajoutés, puis on additionne 4ml de H2O2
(30%) de manière à ce que la température ne dépasse pas 5°C. La solution est
agitée 30 min à 0°C, puis le mélange est hydrolysé avec 30 ml d’eau et 2 ml de
soude (2N).
Le mélange est extrait à
l’acétate d’éthyle. La phase aqueuse est acidifiée jusqu'à pH 3 avec HCl (2N)
et extraite de nouveau à l’acétate d’éthyle. Les phase organiques sont
rassemblées et séchées sur sulfate de
sodium puis évaporées sous vide. 2 mmoles d’une huile translucide sont
obtenues.
Rdt :58%.
IR (cm-1) :
3342 (nNH),
2976 (nCH), 1715 (nC=O acide), 1643 (nC=O BOC)
1H RMN (MeOD) (300MHz) :
1,32 (d, J= 6.8Hz, 3H, NCHCH3) ,
1,45 (s, 9H, (CH3)3C) , 2,05-2,015 (m, 2H, NCHCH3) ,
3,00 (d, J= 10 Hz), 5,20 (s, 1H, NH), 5.55 (m, 1H, CH=), 5.65 (m, 1H,
CH=).
13C RMN (MeOD) (300MHz) : 22 (CH3),
35 (CH2), 40 (CH), 125 (CH=), 139 (CH=),
160 (C=O BOC), (C=O acide).
6) 3-tosyloxypropanol 20
![]()
0,023 mole (4,4g) de
chlorure de tosyl puis 0,024 mole (3,2ml) de triéthylamine sont ajoutés sur
0,16 mole (12,2g) de prop-1,3-diol .
La solution est agitée 1h30
à température ambiante. Le mélange est dilué avec 50ml de dichlorométhane. La
phase organique est lavée avec une solution d’HCl (10%) (3x20ml), puis avec une
solution de NaCl (saturée).
Le produit est purifié sur
colonne de silice (AE/EP 70/30). Le produit est obtenu sous forme d’une huile
jaune (0.022 moles).
rdt=95%
IR (cm-1) :
3418 (nOH), 3066(narCH), 2961,2893 (nCH), 1357 (SO2)
1H RMN (CDCl3) 1,93 ( s,1H, OH) ), 1,88
(q, J=6Hz, 2H, CH2CH2CH2), 2,46 (s, 3H, ArCH3), 3,70
(t, J=7Hz, 2H, OHCH2CH2CH2), 4,18 ( t, J=7 Hz, 2H, CH2OH),
7,45 (d, J=8Hz, 1H, CH méta), 7,79 (d, J=8Hz, 1H, CH ortho).
7) éther tétrahydropyrannique-3-tosyloxypropanol 21
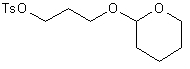
Une quantité catalytique
d’acide paratoluènesulfonique puis 0,02 mole (1,68 g) de dihydropyranne sont
ajoutés en une seul fois sur 0,014 mole (3,2g) du 3-tosylpropanol dissout dans
10 ml de dichlorométhane.
Le mélange est agité 30 min
à température ambiante.
La phase organique est lavée
par une solution saturée d’hydrogénocarbonate de sodium (3x20ml), puis une solution
saturée de chlorure de sodium.
Le mélange est purifié sur
colonne de silice (DCM/MeOH 99/1). 13 mmoles (396 mg) d’une huile jaune sont
obtenues.
Rdt: 95%
IR : 3048 (n CH aromatiques), 2942,2871 (n CH), 1373 (SO2)
1H RMN (CDCl3) (200Mhz): 1,60 (m, 2H,
CHCH2), 1,63 (m, 4H, CH2CH2),
1,93 (q, J=6Hz, OCH2CH2CH2O), 2,46 (s,
3H, CH3), 3,42 (m, 2H, OCH2CH2CH2OSO2),
3,76 (m, 2H, OCH2), 4,16 (m, 2H, CH2OSO2),
4,46 (m, 1H, OCHO), 7,34 (d, J=10Hz, 1H, CH méta), 7,80 (d, J=10Hz, 1H, CH
ortho)
8) Ether tetrahydropyrannique -3-iodopropanol 22
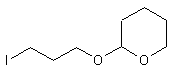
0,03 moles 2,5g de iodure de sodium puis 0,075 moles ( 3,3g
) d’hydrogénocarbonate de sodium sont ajoutés sur 0,015 mole (4,5g) de 7
dissout dans 15 ml d’acétone.
Le mélange est agité pendant
4 h à température ambiante.
Après 4 h la phase organique
est lavée avec une solution saturée de chlorure de sodium, séchée sur sulfate
de sodium puis évaporée sous vide.
Le produit est purifié sur
colonne de silice (DCM/MeOH 98/2). Le produit est obtenu sous forme d’une huile
jaune 0,013 moles (3,4 g)
Rdt : 92%
IR (cm-1) :2973,2935,2872
(nCH), 1199 (dC-O-C), 550 (nC-I).
1H RMN (CDCL3) (200Mhz) : 1,68 (m, 2H, CHCH2),
1,69 (m, 4H, CH2CH2), 2,09 (q, J=7,5Hz, 2H, OCH2CH2CH2I),
3,30 (t, J=7Hz, 2H, ICH2CH2CH2O), 3,48
(m, 2H, OCH2CH2CH2I), 3,84 (m, 2H, CH2O),
4,60 (m, 1H, OCHO)
9) Iodure propanol triphényl phosphonium 23

6 mmoles (1.57g) de
triphénylphosphine sont ajoutées sur 6 mmoles (900 mg) d’éther
tetrahydropyrannique 3-iodopropanol dissout dans 5ml de mesitylène.
Le mélange est porté au
reflux du mesitylène (170°C) pendant 2 h.
Le produit obtenu est
filtré.
Rdt 80%.
IR (cm-1) :3376
(nOH), 3075,3007 (nCH aromatique), 2944,2888,2861
(nCH), 1113 (C-O)
1H RMN (CDCl3) (300MHz) :1,88 (m, 2H, P+CH2CH2CH2),
2,1 (s, 1H, OH), 3,80 (m, 2H, CH2P),
4,18 (m, 1H ,CH2O), 7,82 (m, 15H, CH aromatiques)
10) Synthèse de l’iodure de l’éther tetrahydropyrann de
propyl triphényl phosphonium 19

41 mmoles (0,35g) de
dihydropyranne et une quantité catalytique d’acide paratoluènesulfonique ( 30mg
) sont ajoutés en une seule fois sur 27mmole ( 1,2g ) de 9 en solution
dans le dichloromethane ( 10 ml ).
Le mélange est agité 1 h à
température ambiante puis traité par une solution saturée d’hydrogénosulfate de
sodium et séché sur sulfate de sodium puis évaporé sous vide.
Le produit cristallisé
obtenu est purifié sur colonne de
silice (DCM/MeOH 95/5).
21 mmoles d’une pâte jaune
sont obtenues.
Rdt : 80%
IR (cm-1) : 3376
(n), 3075,3007 (nCHaromatique), 2944,2888,2861 (nCH), 1113 (C-O)
1H RMN (CDCl3) (200MHz) :1,50 (m, 2H,CH2CH2CH2),
1,75 (m, 2H, CH2CH2CHO), 1,95 (m, 2H, CH2CH2O),
3,80 (m, 6H, CH2P+, CH2CHO, CH2CHO),
4,50 (m, 1H ,CHO), 7.82 (m, 15H, CH aromatiques)
11) Synthèse de N-(t-butoxycarbonyl)-5(S)-amino hex-3-ènal
tetrahydropyranyl (E) 18
Sur une solution de 10 2,5 mmoles (800 mg) dans le THF à -60°C est ajouté goutte
à goutte
2 ml de butyllithium. Le mélange est agité 30 min à
-60°C.
Après 30 min est ajouté une solution de 2 2mmoles (360mg) dans le THF,
après l’ addition le mélange est ramené à température ambiante. Le milieu est
agité 2 h à température ambiante.
La solution est versée dans un solution de tartrate
de sodium et tartrate de potassium. La phase aqueuse est extraite à l’AE. La
phase organique est traitée successivement par 3x30ml d’une solution d’HCl 10
%, puis par une solution saturée d’hydrogénocarbonate de sodium et enfin une
solution saturée de chlorure de sodium. La phase organique est séchée sur
sulfate de sodium puis évaporée sous vide.
Le produit est purifié sur colonne de silice ( AE/EP
2/8 ). Le produit est obtenu sous forme d’une huile marron 2 mmoles ( 596 mg ).
Rdt : 45 %
IR (cm-1) : 3320 (n NH), 2923, 2848 (nCH), 1680 (nC=O Boc), 1263 (nC-O), 1069 (nC-O THP)
12) Synthèse de
(S)-N-(t-butoxycarbonyl)-5-aminohex-3-ènoique acide (E) 5
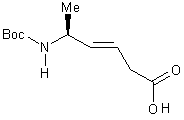
Sur 0,3 mmoles ( 110mg ) de 11 en solution dans l’acétone à 0°C est ajouté goutte à
goutte le réactif de Jones ( 1,92 M ). Le mélange est agité pendant 4h à 0°C
Après 4 h le milieu est dilué avec de l’eau. La
phase aqueuse est extraite par de l’acétate d’éthyle. La phase organique est
lavée par NaOH (2N). La phase aqueuse est acidifié jusqu'à pH 3 par HCl ( 2N )
puis de nouveau extraction à l’acétate d’éthyle. La phase organique est séchée
sur sulfate de sodium, puis évaporée sous vide.
Le produit est obtenu sous forme d’une huile
collante.
Rdt 60 %
IR (cm-1) idem 17
Références
bibliographique
[1] M. M. Hann, P.G. Sammes, J.Chem.Soc., Chem.Comm., (1980), 234-235
[2] A. Spaltenstein, P. A. Carpino, F. Miyake, P. B. Hopkins, Tetrahedron Letters, 1986, 27 (9), 2095-2098
[3] W. J. Thompson, T. J. Tucker, J. E. Schwering,
J. L. Barnes, Tetrahedron Letters,
1990, 31 (47), 6819-6822
[4] J. A. McKinney, D. F. Eppley, R. M. Keenan, Tetrahedron Letters, 1994, 35 (33), 5985-5988
[5] T. Ibuka, H. Yoshizawa, H. Habashita, N. Fujii, Tetrahedron Letters, 1992, 33 (26), 3783-3786
[6] M. M.
Hann, P.G. Sammes, P. D. Kennewell, J. B. Taylor, J.Chem.Soc.Chem.Comm., 1982, 307-314
[7] A. Spaltenstein, P. A. Carpino, F. Miyake, P. B.
Hopkins, J.Org.Chem., 1987, 52,
3759-3766
[8] S. Hanessian, R. Frenette, Tetrahedron letters, 1979, 36, 3391-3394
[9] N.J. Miles, P.G. Sammes, P.D. Kennwell,
R.Westwood, J.Chem.Soc., Perkin Trans 1,
(1985), 2299-2305
[10] J.A. Fehrentz, B. Castro, Synthesis, (1983), 676-678
[11] Y. Zhang, G.B. Schister, J.Org.Chem., (1995), 60,7192-7197
[12] A. Oku, M. Kinugasa, T. Kamada, Chem. Lett., (1993), 165-168
[13] G. Johnston, W.J. Ken, J.S. Scott, J.Chem.Soc., Chem.Com., (1996), 341-342
[14] S.E. Sen, S.L. Roach, J.K. Boggs, G.J. Ewing,
J. Magrath, J.Org.Chem., (1997), 62,
6684-6688
[15] E. Marcantoni, F. Nobuli, J.Org.Chem., (1997), 62, 4183-4184
[16] G. Baudin, Y. Pietrasanta, B. Pucci, Tetrahedron, 1977, 33, 3105-3110
[17] J.C. Stowell, D.R. Keith, Synthesis, (1979), p.132
[18] finkelstein
[19] Evans